E-CAM started in October 2015 and received funding from the European Union’s Horizon 2020 R&I program for a period of 66-months.
Built around the scientific community of the Centre Européen de Calcul Atomique et Moléculaire (CECAM), E-CAM was coordinated by CECAM Headquarters (HQ) at EPFL, and was a partnership of 14 CECAM Nodes, 4 PRACE centres and one Centre for Industrial Computing (Hartree Centre).
The present final report provides an overview of the project results during the EU funding period, and includes:
project highlights
overview of the project results in each Work-Package (WP)
Computation based methods play a growing role in all stages of accelerated medicine pipelines responding to industry challenges of drug substance development.
Abstract
APC was created In 2011 by Dr. Mark Barrett and Prof. Brian Glennon of the University College Dublin School of Chemical and Bioprocess Engineering with a mission to harness state of the art research methods & know-how to accelerate drug process development. Since then it has grown organically partnering with companies across the world, large and small, to bring medicines to market at unprecedented speed. Computation-based methods play a growing role in all stages of its medicine pipeline as explained by Dr. Jacek Zeglinski in this E-CAM interview on the challenges to Industry of drug substance development.
What is APC?
APC, the company I work for stands for Applied Process Company, and we are based in South County Dublin in Ireland, actually, just half an hour walk from the Irish Sea. APC is a global company, although it’s perhaps not very big, it is an impactful company because we collaborate with a range of big pharma companies across the world, both big and small. We provide services to them to help accelerate the development of active pharmaceutical ingredients (APIs), both small molecules, and biomolecules. We work in a number of domains of product development, starting from early-stage development through to optimization up to scale up and technology transfer.
Can you tell us how you got involved with APC?
I learned about APC in 2012, while working as a postdoc at the University of Limerick. At that time APC was a newly established start up company. Some time later two friends from my research group joined APC, so very soon I had first-hand opinion of the company. It appeared that the superior company culture and the dive-deep research focus were sufficiently strong arguments for me to join APC in early 2019.
What challenges does APC focus on, say in the context of drug development?
When we refer to drugs or drug substances, we have in mind the powder forms that are the basis for making tablets. Usually, they are crystalline but sometimes they are in amorphous form. There are a number of challenges we face. The poor solubility of active ingredients is probably the most important. Until recently people would have considered APIs to be small molecules, but nowadays they are getting larger and larger, up to 1000 grams per mole in molecular weight. They are also quite flexible and complex and that actually brings challenges related to their solubility. Most of the APIs we are handling have poor solubility in a range of solvents. That would translate to insufficient bio-availability, and make it difficult to manufacture those APIs with high productivity (i.e. producing low yield and throughput) if we could not solve the problem of solubility.
The second challenge we face is polymorphism. Crystalline materials can exist in a number of different crystal forms, those forms are interrelated in terms of stability, some of them are metastable and the most stable ones and not always easy to obtain. Sometimes one gets unwanted solvates or hydrates at the start of the development process when screening is being done in a range of different solvents. The third issue relates to particle size and shape, which can cause poor processability, filter-ability, flow-ability, compressibility, and difficulty to form tablets. Particles that are too large can have poor or inconsistent dissolution rates and bioavailability. Agglomeration is another challenge, and consequent difficulties to remove impurities, particularly when they are hidden in the voids within agglomerates.
What computational approaches do you use?
In our work in optimizing processes, we mainly do a lot of experiments but we also use computational methods. I will briefly highlight the latter, starting with our typical workflow for solvent selection for crystallization processing.
Solubility Predictions for Crystallisation Processes
Computational tools allow us to screen many more solvents than would be possible using experimentation alone. The workflow usually starts with 70 solvents, which are screened computationally regarding their solubility temperature dependence, the propensity of forming solvates or hydrates, and their utility for impurity purging, ending up with less than 10 promising solvents for experimental validation. This computational screening which is done in two to three days would take several weeks or even months were experiment used instead. We then check for the crystal form, for purity, and some other parameters that allow us to narrow down our candidates to two, three, or even one solvent for small-scale crystallisation experiments. The most optimum solvent system is further used to develop the full process followed by a final scale-up demonstration. So as you can see that computational part is a very important piece of this workflow and it really aids the solvent selection process. Regarding software, we are using mainly two platforms from Biovia, one is COSMO-therm (combined with a DFT-based program – TURBOMOLE) and the other one is Materials Studio. With COSMO we predict solubilities, and also the propensity for solvate/cocrystal and even salt formation. With Materials Studio, we search for the low energy conformations of molecules and use those conformations for solubility prediction using COSMO, so the two software codes work together. With Materials Studio, we also do lattice and cohesive energy calculations, so as to estimate thermodynamic stability of polymorphs, or find imperfections in the crystal lattice or predict the shape of crystals. We are constantly developing these capabilities and trying to do more predictive modeling. Looking at the molecular complexity of the future medicines we handle, we clearly see that there is a real need to better understand the solid-state features of those APIs, which very often are multicomponent materials, e.g. cocrystals, salt-cocrystal-hydrate hybrids etc.
How do the computational solubility predictions compare with experiment?
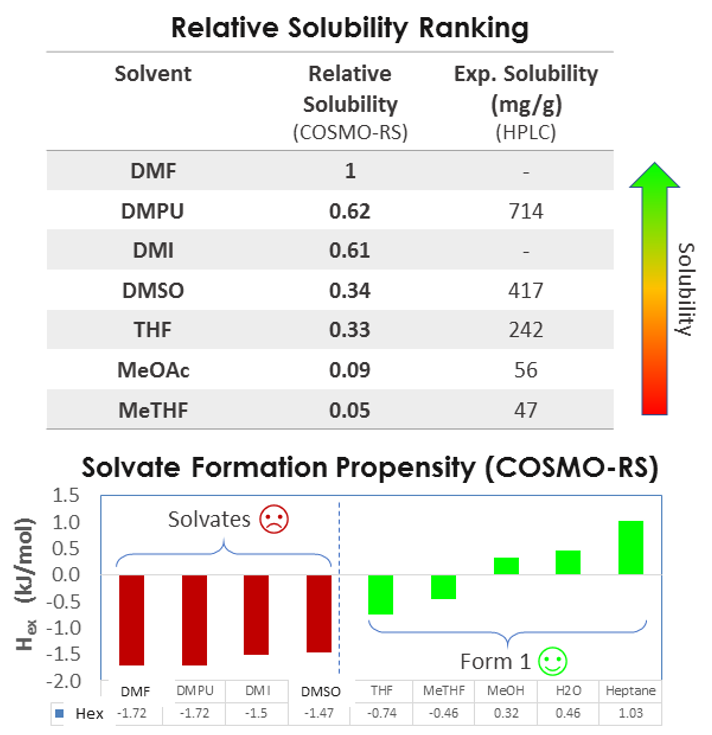
I would like to show how relative solubility predictions performed with COSMO compare with experiments for a medium-sized API molecule (500g/Mol) in a variety of solvents. As you can see there’s a good match between the rank order of solubility.
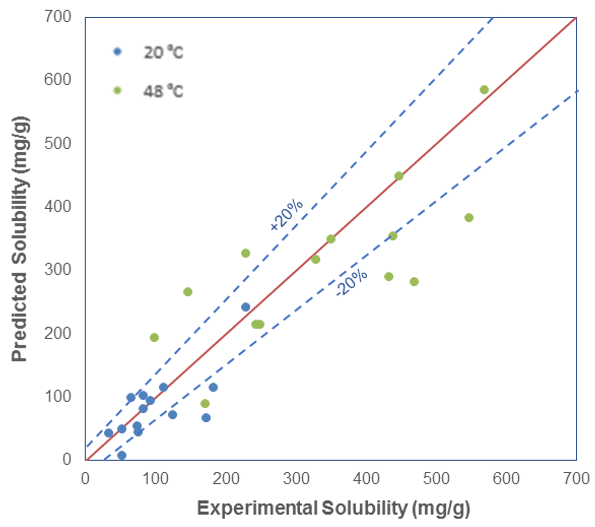
So we can predict relative solubility and, in addition, we can compute the propensity for solvate formation, that is the enthalpy of mixing, which is related to the interaction between solvent and solute. More negative values indicate exothermic events and stronger interactions, and more positive values indicate endothermic events and unfavorable mixing. So the case study presented was a very successful validation of this approach. In addition to relative solubility, we can predict absolute solubility and compare it with experiment, for example at two different temperatures, 20 and 48 °C.
As you can see, for some of the systems, the solubility is predicted very well, but for most systems, the computational and experimental results are within 20% of each other. At lower temperatures, the agreement is very good, but at higher temperatures, we see the predictions are not as accurate.
In addition to single solvents, we can predict solubility in solvent mixtures. This is a very useful application because, for many APIs, there is no single neat solvent that can be identified. In such cases, there is often a solvent mixture that can be identified which gives rise to good solubility for the API. For example, in the binary solvent mixture of water and ethyl acetate, certain solvent ratios give rise to good solubility for a variety of solutes. We can also have ternary, quaternary, and higher mixtures of solvents studied computationally.
How important is the stability of different conformers of an API in a solvent?
Conformational Aspects in Solubility Predictions
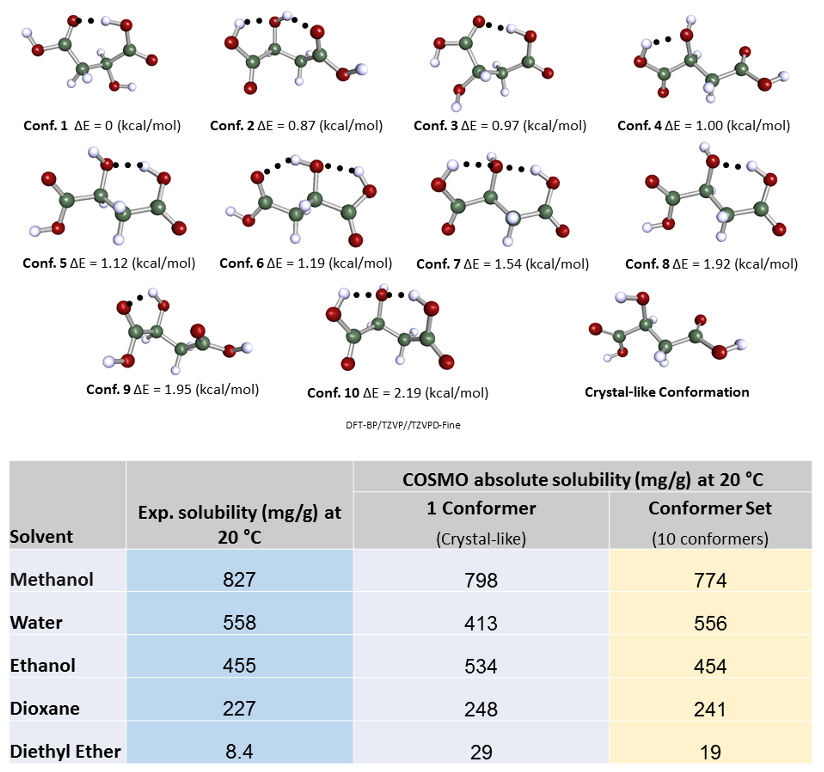
This is perhaps more fundamental, and interesting. I will show an example of solubility predictions for malic acid, and focus on different conformations. Malic acid can make a number of intra-molecular hydrogen bonds and that will be reflected in different relative conformational energies. There’s a hypothesis that regarding solubility predictions, the lowest energy conformations should be dominant because this is the confirmation that should be the most probable and most frequently occurring in solution. However, we find that this is not always the case. We can compare the predicted values of solubilities for five different solvents with experimental values for the crystal-like molecular conformation obtained in vacuum with DFT using COSMO and ten other low energy conformers identified based on their relative energetic stability.
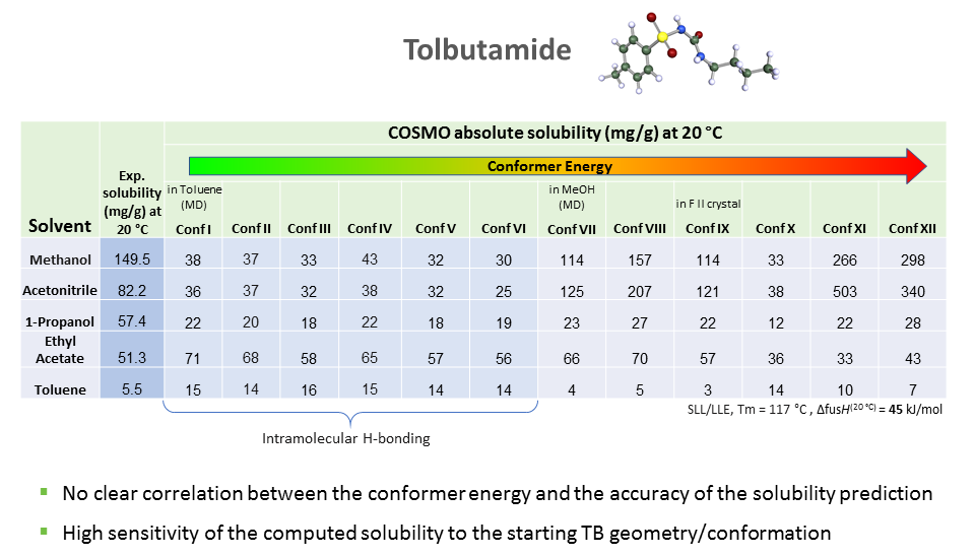
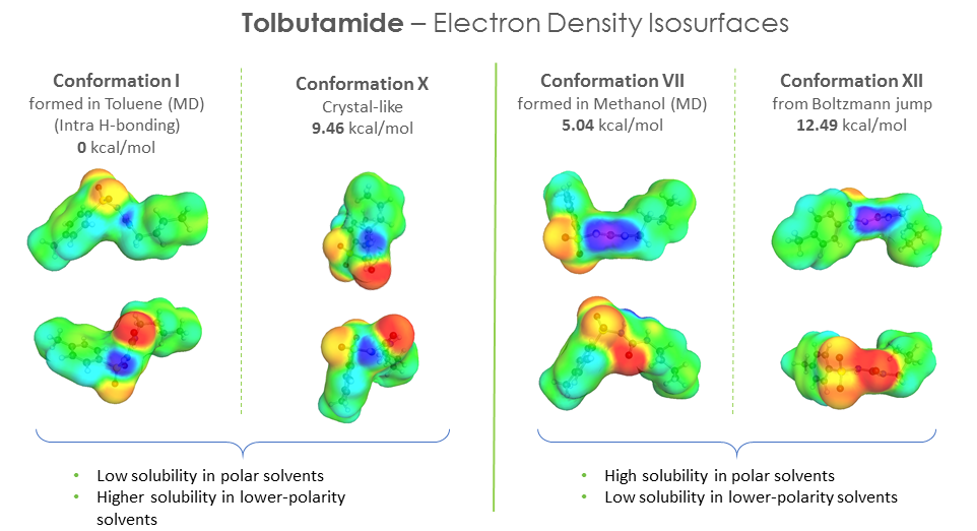
As we can see the predicted solubility compare fairly well with experiment, particularly in terms of relative ranking of solubility, as this allows the best solvent to be chosen. We see that the comparison using ten conformers is slightly better for predicting absolute solubility. However, it is not always the case that we have such good agreement for solubilities between prediction and experiment. For molecules that are larger and more flexible, the challenge increases. Another case we have studied is tolbutamide. In polar solvents such as methanol, the molecule should not have an intra-molecular hydrogen bond conformation, but in non-polar or low polarity solvents such as toluene, it should. We generated a variety of conformations some of which had intra-molecular hydrogen bonds and predicted the solubilities using those different conformations having different relative energies.
The hypothesis that the lower the energy of the conformer, the better its solubility does not apply in this case. There is no clear correlation between conformer energy and the accuracy of the solubility predicted when compared with the experiment for tolbutamide.
We also looked at the electron density iso-surfaces for the different conformers and found a correlation between the polar “domains” in the distribution (local concentration of positive and negative charge distributions) and the solubility in polar solvents and vice-versa for non-polar solvents – where the distribution of positive and negative charges were more scattered and less pronounced. It would be nice to quantify this and explore it further.
How do you predict the morphology of APIs and how well do they compare with experiments?
Crystal Shape/Morphology Prediction of Pharmaceutical Compounds
In our projects, before starting any experiments we try to predict as many properties as possible, including shape/morphology of crystals of a particular API. The predictions are made in vacuum, and very often reflect the experimental morphology. There are some exceptions, but in most cases, we find good agreement. We use an attachment energy theory which assumes that crystal growth is proportional to the attachment energy, i.e. the energy released when a slice of crystal is added to a particular facet (or lattice direction). The higher the attachment energy on the facet, the faster the growth in that direction. Based on that, we can predict the shape of the crystalline particle and the intrinsic propensity for its growth in particular directions, which will also give us the theoretical aspect ratio. It turns out that this is often related to the relative polarity of different facets; basically the number of polar atoms at different surfaces. Usually, the higher the polarity, the stronger the hydrogen bonding, and the faster will be the growth rate in that direction. However, there can be exceptions to that. For example, for molecules that are elongated and somewhat flat like Clofazimine, there are no intermolecular hydrogen bonds that occur, and molecular packing is more important. Ideally, we would like to predict the effect of the solvent on the crystal shape. Sometimes we are able to find a correlation between the surface chemistry and the morphology we are trying to predict and the impact of a particular solvent type. For example, some systems have high polarity charge distributions on certain facets, and in such cases, if we use polar-protic solvents, they would preferentially interact with polar atoms on that facet and inhibit crystal growth there. On the other hand, they could allow growth along low polarity directions. We found some experimental preliminary evidence of this, but it still has to be confirmed. We are trying to use this sort of analysis to predict which solvent will generate the desired morphology. We also explore more complex modelling approaches, including implicit and explicit solvent effects on crystal morphology and our modelling work done in this context is promising. I would like to thank the CEO of APC, Dr. Mark Barret for the opportunity to do this work, and my co-authors, Dr. Marko Ukrainczyk and Prof. Brian Glennon.
The transformation of a beautiful idea born via simulation into a commercial opportunity is recognised as a disruptive technology. At the heart of this ongoing story is advanced simulation using massively parallel computation, rare-event methods and genetic engineering.
Proof of concept : recognition as a disruptive technology
Author: Donal Makernan, University College Dublin, Ireland
Last week I received an email asking if I would be willing to accept the ‘2021 NovaUCD Licence of the Year Award’ for the licence of the disruptive molecular switch platform technology to a US based company with an initial application as a point-of-care medical diagnostic for COVID-19 and influenza. Of course I said yes, and since then received a beautiful statue of a metal helix mounted on a black marble plinth via courier (displayed on the right). It is nice that our work gets this sort of recognition given all of the effort it has taken to get to this point. In my last blog post I wrote of the first steps towards commercialization of our technology. Since then, everything has intensified. The company funding this research collaboration with University College Dublin has now over 20 people in the USA dedicated to its commercialization – including old hands hired from well known immuno-diagnostic and pharmaceutical companies, medical doctors, engineers and sales-persons. On our side, our team has grown and now includes two software-engineers/simulators trained in part through E-CAM while they were studying theoretical physics, and four molecular biologists. In addition, contract research and manufacturing organizations are also now being engaged so as to be ready for clinical testing and scale-up when we have fully optimized our diagnostic sensors for COVID 19. Hard to believe it is only one year since we met the key commercial people. We continue to simulate various forms of the sensor so as to optimize its performance and commercialization, and for that HPC resources from PRACE partners from Ireland (ICHEC), Switzerland (CSCS) and Italy (Cineca) have been of huge help. We also are dedicating a lot of effort in software development so as to speed up our ability to estimate free energy properties such as binding affinities, which turn out to be much tricker than one might expect when proteins are very large, such as between antibodies and target antigens such as the COVID 19 spike protein. That methodology and software arose from an E-CAM pilot project – and would appear to have a potential utility way beyond our first expectations. The E-CAM Centre of Excellence grant from the EU will be finished soon (31st March). Hopefully it will emerge soon again.
The development of enhanced sampling methods to investigate rare but important events has always been a focal point in the molecular simulation field. Such methods often rely on prior knowledge of the reaction coordinate. However, the search for this reaction coordinate is at the heart of the rare event problem. Transition path sampling (TPS) circumvents this problem by generating an ensemble of dynamical trajectories undergoing the activated event. The reaction coordinate is extracted from the resulting path ensemble using variants of machine learning, making it an output of the method instead of an input. Over the last 20 years, since its inception, many extensions of TPS have been developed. Perhaps surprisingly, large‐scale TPS simulations on complex molecular systems have become possible only recently. Other important developments include the transition interface sampling (TIS) methodology to compute rate constants, the application to multiple states, and adaptive path sampling. The development of OpenPathSampling and PyRETIS has enabled easy and flexible use and implementation of these and other novel path sampling algorithms. In this progress report, a brief overview of recent developments, novel algorithms, and software is given. In addition, several application areas are discussed, and a future outlook for the next decade is given.
This module sets up the European Environment for Scientific Software Installations (EESSI) for use in GitHub Workflows.
The European Environment for Scientific Software Installations (EESSI) is a collaboration between a number of academic and industrial partners in the HPC community to set up a shared stack of scientific software installations to avoid the installation and execution of sub-optimal applications on HPC resources. The software stack is intended to work on laptops, personal workstations, HPC clusters and in the cloud, which means the project will need to support different CPUs, networks, GPUs, and so on.
The EESSI project is supported by E-CAM, and forms the basis of the software stack used within the LearnHPC project (which is also supported by E-CAM).
EESSI can be leveraged in continuous integration (CI) workflows to easily provide the dependencies of an application. With this module we create a GitHub Action for EESSI so that it can be used within a projects CI on GitHub. By using the Action, you can use environment modules to resolve the dependencies of your application in highly predictable and reproducible way. This includes state-of-the-art compilers, MPI runtimes and mathematical libraries.
This module describes the work done in E-CAM in cooperation with the HemeLB code from the CompBioMed Centre of Excellence.
HemeLB is a high performance lattice-Boltzmann solver optimised for simulating blood flow through sparse geometries, such as those found in the human vasculature. The code is used within the CompBioMed HPC Centre of Excellence H2020 project and is already highly optimised for HPC usage. Nevertheless, in an E-CAM workshop on the load balancing library ALL hosted at the Juelich Supercomputing Centre, a cooperation was set up in order to analyse and test whether the use of ALL could improve the existing scalability of the code.
ALL was designed to work with particle codes, therefore it was interesting to apply the library to a lattice-Boltzmann solver, which usually is not particle-based. The different grid points of the solution grid were designated as particles and since each of the grid-points already was assigned a workload, the sum of grid-point workloads could be used as domain work load.
As a result, it was demonstrated that the domain compositions provided by ALL show a better theoretical load distribution. Tests to check if this translates into better code performance are inconclusive as yet, due to hardware related issues on the testing platforms. However, these are currently under further investigation, and more definitive results about the performance of the ALL-provided domain decompositions can be expected in the near future. The results were part of an article about HemeLB, which was published in 2020[1] .
Towards blood flow in the virtual human: efficient self-coupling of HemeLB J. W. S. McCullough, R. A. Richardson, A. Patronis, R. Halver, R. Marshall, M. Ruefenacht, B. J. N. Wylie, T. Odaker, M. Wiedemann, B. Lloyd, E. Neufeld, G. Sutmann, A. Skjellum, D. Kranzlmüller and P. V. Coveney Interface Focus2020, 11: 20190119 DOI: http://dx.doi.org/10.1098/rsfs.2019.0119 (open access)
To further expand the portfolio of activities targeted at industrialists, E-CAM has established a series of new events targeted at training interested industrial researchers on the simulation and modelling techniques implemented in specific codes and in the direct use of this software for their industrial applications.
The first event of this series will focus on the area of meso- and multiscale simulations and on the flagship code DL_MESO:
In this workshop we will introduce DL_MESO: a software package for mesoscale simulations. Usage of the software will be gradually presented, starting with tutorials based on theoretical background and following up with hands-on sessions. We will focus on the Dissipative Particle Dynamics (DPD) methodology, exploring the different capabilities of DL_MESO_DPD via practical examples that reflect daily industrial challenges.
DL_MESO has been used for a wide range of problems of both scientific and industrial interest. The code is used, for example, in projects with Unilever, Syngenta and Infineum – to develop DPD parameterisation strategies and simulation protocols to predict important properties of newly-devised surfactant-based formulations; with IBM Research Europe – to model nanofluidic multiphase. The code developers themselves will provide the training. The event is co-organized by Formeric, a company that helps industrial users to study their own formulated projects, primarily by developing a software platform to make it easier for them to access DPD simulations and modelling tools.
As part of the event, UKRI STFC offers a 6-month one seat free licence of DL_MESO 2.7 to be used soon after the end of the event, which will help testing the software.
Don’t miss this opportunity to be trained by the experts on the methods and on the codes themselves! Register for event at
Our last Extended Software Development Workshop (ESDW) took place on the 18th-22nd January[1], and given its length (5 days) and it’s nature (theory and hands-on training sessions) it was a real success! “The workshop went very well, participants seem to have enjoyed and they lasted until the end !”, said organiser Jony Castagna, computational scientist and E-CAM programmer at UKRI STFC Daresbury Laboratory. The event, organised at the CECAM-UK-DARESBURY Node[2], focused on HPC for mesoscale simulation, and aimed at introducing participants to Dissipative Particle Dynamics (DPD) and the mesoscale simulation package DL_MESO [3] (DL_MESO_DPD). DL_MESO is developed at UKRI STFC Daresbury by Michael Seaton, computational chemist at Daresbury and also an organiser of this event.
Another component of this workshop was parallel programming of hybrid CPU-GPU systems. In particular, DL_MESO has recently been ported to multi-GPU architectures[4] and runs efficiently up to 4096 GPUs, an effort supported by E-CAM (thank you Jony!). Part of this workshop was dedicated to theory lectures and hands-on sessions on GPU architectures and OpenACC (NVidia DLI course) given by Jony, which is an NVidia DLI Certified Instructor. He said “The intention is not only to port mesoscale solvers on GPUs, but also to expose the community to this new programming paradigm, which they can benefit from in their own fields of research”.
All sessions in this ESDW were followed by discussions and hands-on exercises. Organisers were supported by another STFC colleague and former E-CAM post-doc Silvia Chiacchiera. One of the participants wrote “Thank you so much for your effort. This workshop will cause a significant shift in my thinking and approach”.
21 people registered for to the event; but by the third day there were only 9… from which 5 lasted until the last session! A picture taken from the last session talks by itself 🙂
Do you want to join our next training event ? Check out our programme :
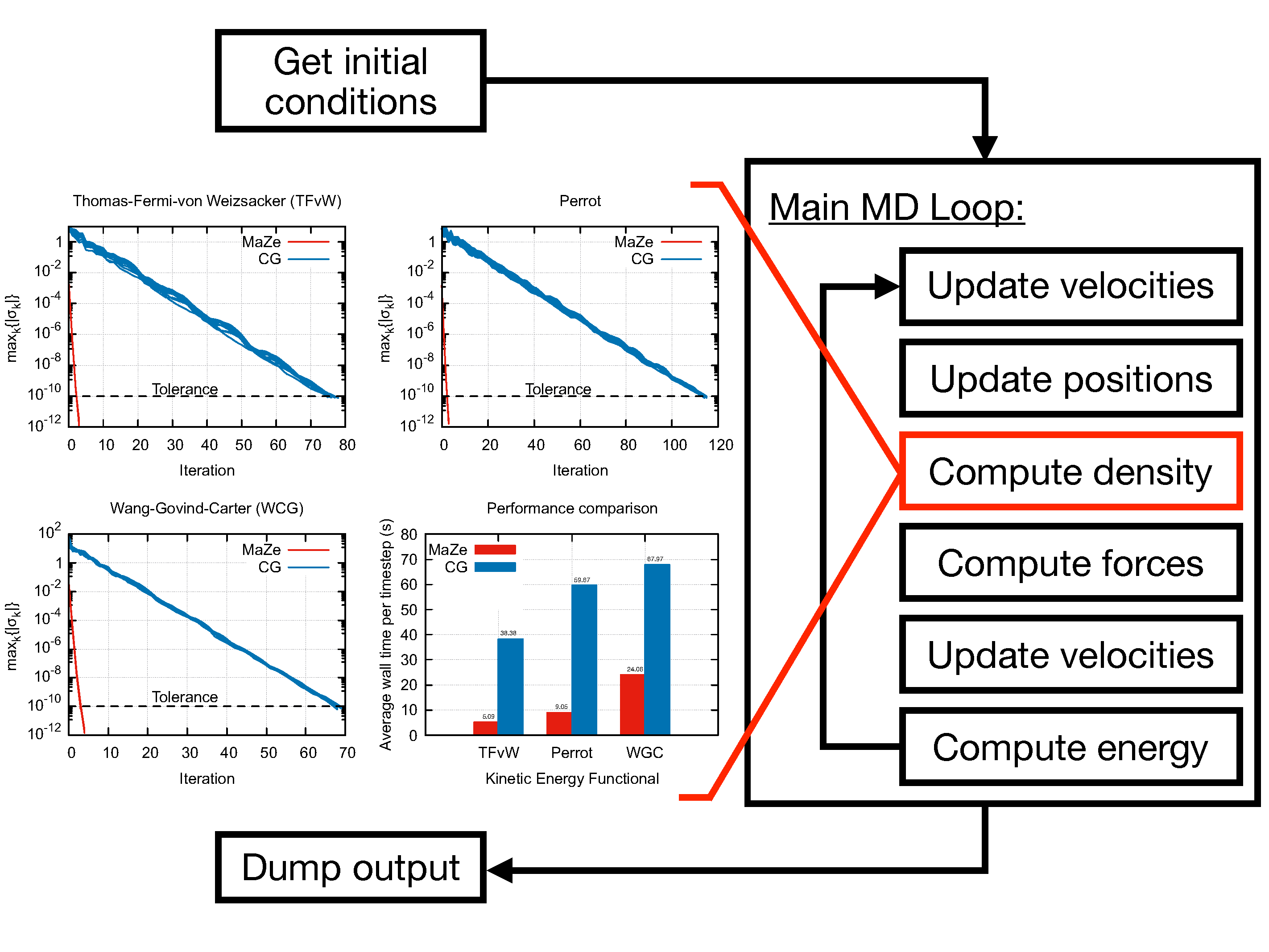
The program performs Orbital-Free Density Functional Theory Molecular Dynamics (OF-DFT-MD) using the Mass-Zero (MaZe) constrained molecular dynamics approach described in [1].
This method enforces, at each time step, the Born-Oppenheimer condition that the system relaxes instantaneously to the ground state through the formalism of massless constraints. The adiabatic separation between the degrees of freedom is enforced rigorously, and the algorithm is symplectic and time-reversible in both physical and additional set of degrees of freedom.
The computation of the electronic density is carried out in reciprocal space through a plane-waves expansion so that the mass-zero degrees of freedom are associated to the Fourier coefficients of the electronic density field. The evolution of the ions is performed using Velocity-Verlet algorithm, while the SHAKE algorithm is used for computation of the additional degrees of freedom. The code can sample the NVE and the NVT ensemble, the latter through a Langevin thermostat.
The code was optimised to run on HPC machines, as explained in the software documentation. The proposed optimisations allow a reduction of the execution time by roughly 50% compared to the original version of the code.
Caption: MaZe optimisation of the electronic density at each nuclear step along an orbital-free DFT Born–Oppenheimer trajectory. Very high speed of convergence is achieved by interpreting the optimisation as a constraint solved via an original implementation of the SHAKE algorithm. The number of iterations needed to converge the electronic density and the time per time step for MaZe (red) and standard conjugate gradients (blue) are compared for the indicated kinetic energy functionals (G_c is the energy cut-off).
Practical application
The code is intended for condensed matter physicists and for material scientists and it can be used for various purposes related to the subject. Even though some analysis tools are included in the package, the main goal of the software is to produce particles trajectories to be analysed in post-production by means of external software.
MaZe implements the orbital-free formulation of density functional theory, in which the optimisation of the energy functional is performed directly in terms of the electronic density without use of Kohn-Sham orbitals. This feature avoids the need for satisfying the orthonormality constraint among orbitals and allows the computational complexity of the code to scale linearly with the dimensionality of the system. The accuracy of the simulation relies on the choice of the kinetic energy functional, which has to be provided in terms of the electronic density alone.
Documentation and source code
The complete documentation is at this location. The source code is available from the E-CAM Gitlab under the MaZe project (software is under embargo until publication leveraging the developments is achieved. Contact code developers or info@e-cam2020.eu for more information.)
References
[1] Sara Bonella, Alessandro Coretti, Rodolphe Vuilleumier, Giovanni Ciccotti, “Adiabatic motion and statistical mechanics via mass-zero constrained dynamics”, Phys. Chem. Chem. Phys.2020, 22, 10775-10785 DOI: 10.1039/D0CP00163E Pre-print version (open access): https://arxiv.org/abs/2001.03556