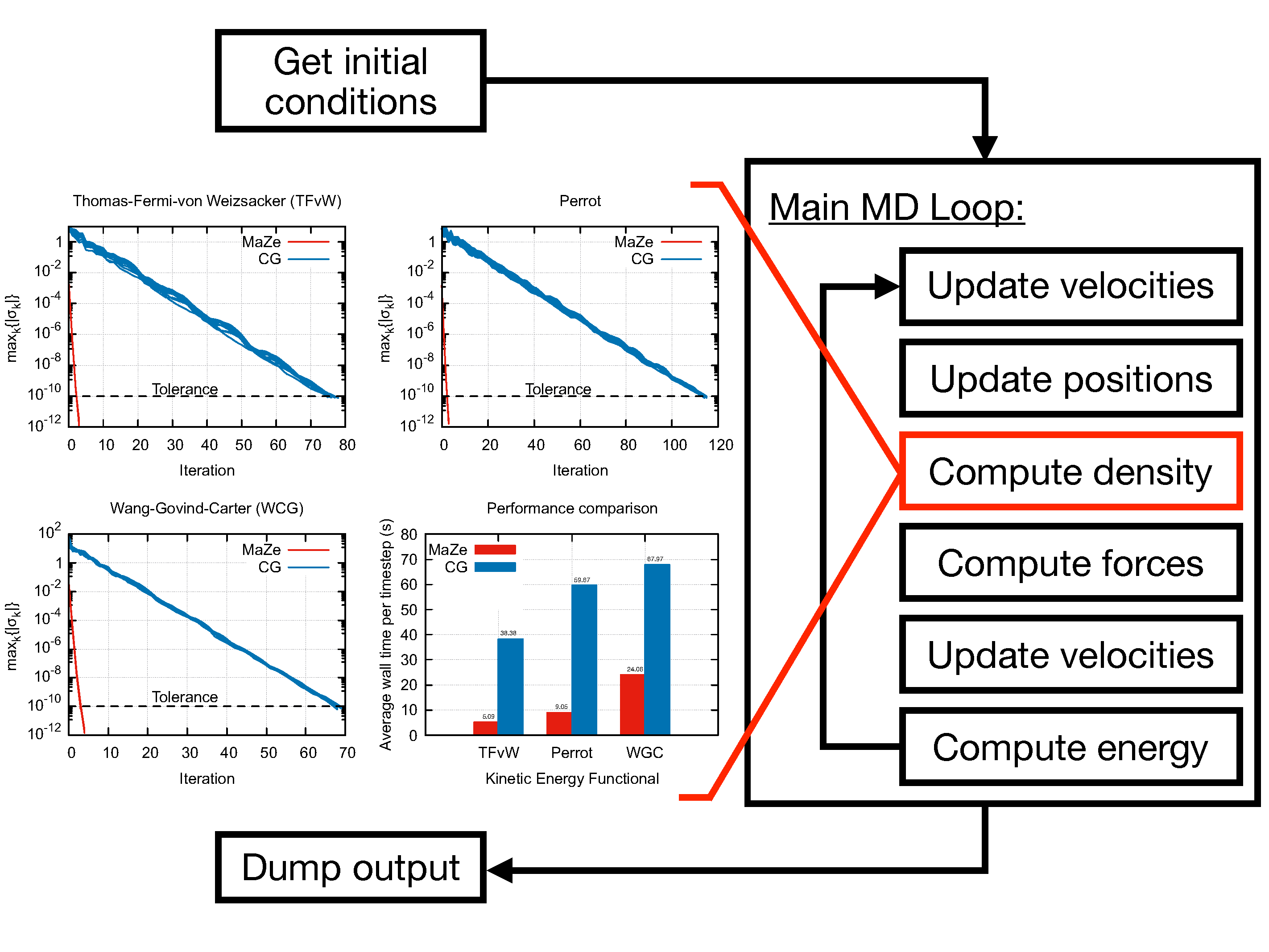
The program performs Orbital-Free Density Functional Theory Molecular Dynamics (OF-DFT-MD) using the Mass-Zero (MaZe) constrained molecular dynamics approach described in [1].
This method enforces, at each time step, the Born-Oppenheimer condition that the system relaxes instantaneously to the ground state through the formalism of massless constraints. The adiabatic separation between the degrees of freedom is enforced rigorously, and the algorithm is symplectic and time-reversible in both physical and additional set of degrees of freedom.
The computation of the electronic density is carried out in reciprocal space through a plane-waves expansion so that the mass-zero degrees of freedom are associated to the Fourier coefficients of the electronic density field. The evolution of the ions is performed using Velocity-Verlet algorithm, while the SHAKE algorithm is used for computation of the additional degrees of freedom. The code can sample the NVE and the NVT ensemble, the latter through a Langevin thermostat.
The code was optimised to run on HPC machines, as explained in the software documentation. The proposed optimisations allow a reduction of the execution time by roughly 50% compared to the original version of the code.
Caption: MaZe optimisation of the electronic density at each nuclear step along an orbital-free DFT Born–Oppenheimer trajectory. Very high speed of convergence is achieved by interpreting the optimisation as a constraint solved via an original implementation of the SHAKE algorithm. The number of iterations needed to converge the electronic density and the time per time step for MaZe (red) and standard conjugate gradients (blue) are compared for the indicated kinetic energy functionals (G_c is the energy cut-off).
Practical application
The code is intended for condensed matter physicists and for material scientists and it can be used for various purposes related to the subject. Even though some analysis tools are included in the package, the main goal of the software is to produce particles trajectories to be analysed in post-production by means of external software.
MaZe implements the orbital-free formulation of density functional theory, in which the optimisation of the energy functional is performed directly in terms of the electronic density without use of Kohn-Sham orbitals. This feature avoids the need for satisfying the orthonormality constraint among orbitals and allows the computational complexity of the code to scale linearly with the dimensionality of the system. The accuracy of the simulation relies on the choice of the kinetic energy functional, which has to be provided in terms of the electronic density alone.
Documentation and source code
The complete documentation is at this location. The source code is available from the E-CAM Gitlab under the MaZe project (software is under embargo until publication leveraging the developments is achieved. Contact code developers or info@e-cam2020.eu for more information.)
References
[1] Sara Bonella, Alessandro Coretti, Rodolphe Vuilleumier, Giovanni Ciccotti, “Adiabatic motion and statistical mechanics via mass-zero constrained dynamics”, Phys. Chem. Chem. Phys.2020, 22, 10775-10785 DOI: 10.1039/D0CP00163E Pre-print version (open access): https://arxiv.org/abs/2001.03556
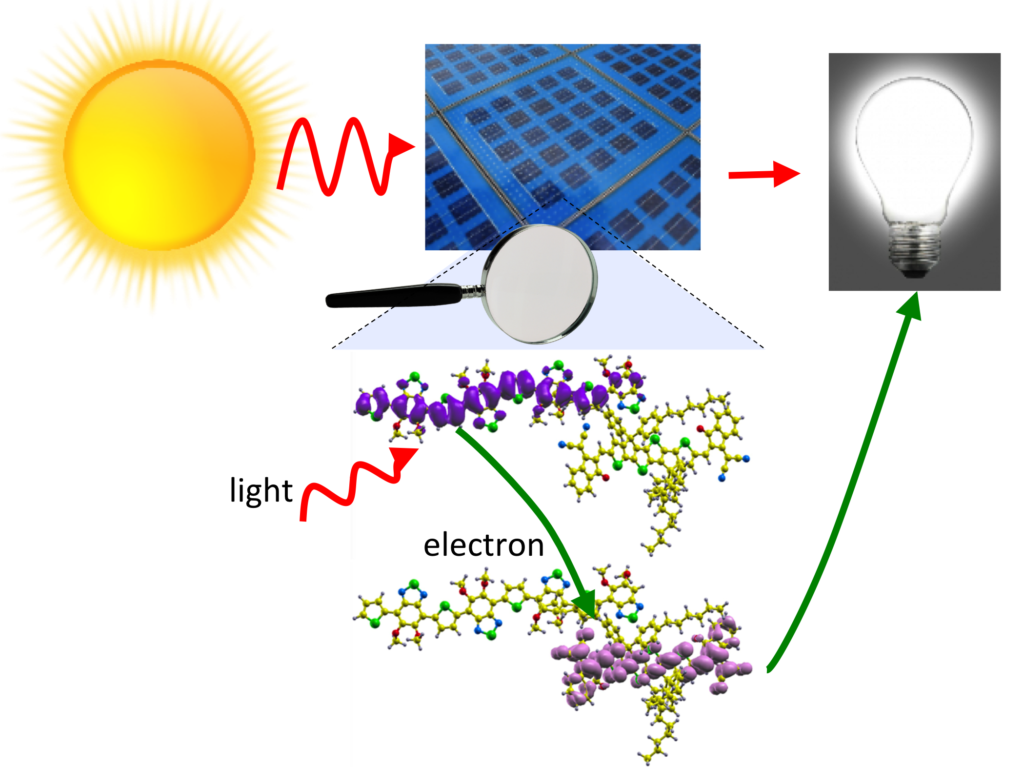
The need to find easily renewable and environmentally friendly energy sources alternative to the traditional fossil fuels is nowadays a global quest. The solar energy is a promising candidate and organic solar cells (OSCs) have attracted attention. In this collaboration with Merck, E-CAM scientists have used electronic structure calculations to study how a key magnitude – the HOMO-LUMO band gap – changes with respect to the molecular disposition of the donor-acceptor molecule pair.
In a recent paper[1], researchers from the Centres of Excellence E-CAM[2] and MaX[3], and the centre for Computational Design and Discovery of Novel Materials NCCR MARVEL[4], have proposed a new procedure for automatically generating Maximally-Localised Wannier functions (MLWFs) for high-throughput frameworks. The methodology and associated software can be used for hitherto difficult cases of entangled bands, and allows the electronic properties of a wide variety of materials to be obtained starting only from the specification of the initial crystal structure, including insulators, semiconductors and metals. Industrial applications that this work will facilitate include the development of novel superconductors, multiferroics, topological insulators, as well as more traditional electronic applications.
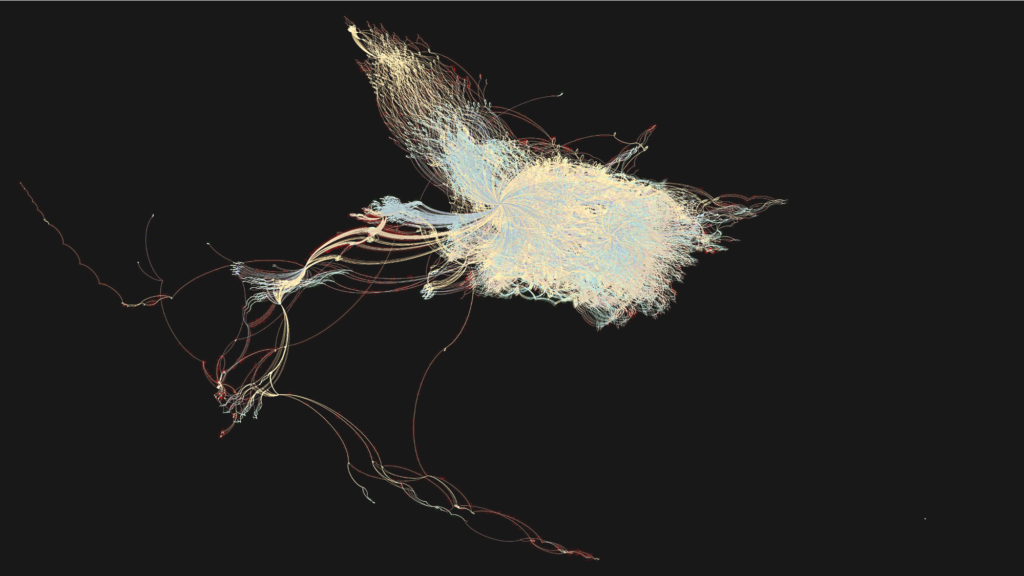
Graphical representation of all data and calculations run in the project and their interconnections (provenance), as tracked automatically by AiiDA in the form of a directed acyclic graph (image credits: G. Pizzi)
Challenge/context
Predicting the properties of complex materials generally entails the use of methods that facilitate coarse grained perspectives more suitable for large scale modelling, and ultimately device design and manufacture. When a quantum level of description of a modular-like system is required, this can often be facilitated by expressing the Hamiltonian in terms of a localised, real-space basis set, enabling it to be partitioned without ambiguity into sub-matrices that correspond to the individual subsystems. Maximally-localised Wannier functions (MLWFs) are particularly suitable in this context. However, until now generating MLWFs has been difficult to exploit in high-throughput design of materials, without the specification by users of a set of initial guesses for the MLWFs, typically trial functions localised in real space, based on their experience and chemical intuition.
Solution
E-CAM[2] scientist Valerio Vitale and co-authors from the partner H2020 Centre of Excellence MAX[3] and the Swiss based NCCR MARVEL [4] in a recent article[1] look afresh at this problem in the context of an algorithm by Damle et al[5], known as the selected columns of the density matrix (SCDM) method, as a method to provide automatically initial guesses for the MLWF search, to compute a set of localized orbitals associated with the Kohn–Sham subspace for insulating systems. This has shown great promise in avoiding the need for user intervention in obtaining MLWFs and is robust, being based on standard linear-algebra routines rather than on iterative minimisation. In particular, Vitale et al. developed a fully-automated protocol based on the SCDM algorithm in which the three remaining free parameters (two from the SCDM method, plus the choice of the target dimensionality for the disentangled subspace) are determined automatically, making it thus parameter-free even in the case of entangled bands. The work systematically compares the accuracy and ease of use of standard methods to generate localised basis sets as (a) MLWFs; (b) MLWFs combined with SCDM’s and (c) using solely SCDM’s; and applies this multifaceted perspective to hundreds of materials including insulators, semiconductors and metals.
Comparison between Wannier-interpolated valence bands (red lines) and the full direct-DFT band structure (black lines), for 150 different materials. The direct and interpolated band structures are essentially indistinguishable (image credits: G. Pizzi)
Benefit
This is significant because it greatly expands the scope of materials for which MLWFs can be generated in high throughput studies and has the potential to accelerate the design and discovery of materials with tailored properties using first-principles high-throughput (HT) calculations, and facilitate advanced industrial applications. Industrial applications that this work will facilitate include the development of novel superconductors, multiferroics, topological insulators, as well as more traditional electronic applications.
Background information
This module is a collaboration between the E-CAM and MaX HPC centres of excellence, and the NCCR MARVEL.
In SCDM Wannier Functions, E-CAM has implemented the SCDM algorithm in the pw2wannier90 interface code between the Quantum ESPRESSO software and the Wannier90 code. This was done in the context of an E-CAM pilot project at the University of Cambridge. Researchers have then used this implementation as the basis for a complete computational workflow for obtaining MLWFs and electronic properties based on Wannier interpolation of the Brillouin zone, starting only from the specification of the initial crystal structure. The workflow was implemented within the AiiDA materials informatics platform (from the NCCR MARVEL and the MaX CoE) , and used to perform a HT study on a dataset of 200 materials.
Source Code
See the Materials Cloud Archive entry. A downloadable virtual machine is provided that allows to reproduce the results of the associated paper and also to run new calculations for different materials, including all first-principles and atomistic simulations and the computational workflows.
Bibliography
[1] Automated high-throughput Wannierisation, Valerio Vitale, Giovanni Pizzi, Antimo Marrazzo, Jonathan R. Yates, Nicola Marzari and Arash A. Mostofi, Nature Computational Materials (2020)6:66 ; https://doi.org/10.1038/s41524-020-0312-y
[5] Compressed Representation of Kohn−Sham Orbitals via Selected Columns of the Density Matrix , Anil Damle, Lin Lin, and Lexing Ying, J. Chem. Theory Comput. 2015, 11, 1463−1469 https://pubs.acs.org/doi/10.1021/ct500985f
First-principles electronic structure calculations are very widely used thanks to the many successful software packages available. Their traditional coding paradigm is monolithic, i.e., regardless of how modular its internal structure may be, the code is built independently from others, from the compiler up, with the exception of linear-algebra and message-passing libraries. This model has been quite successful for decades. The rapid progress in methodology, however, has resulted in an ever increasing complexity of those programs, which implies a growing amount of replication in coding and in the recurrent re-engineering needed to adapt to evolving hardware architecture. The Electronic Structure Library (ESL) was initiated by CECAM to catalyze a paradigm shift away from the monolithic model and promote modularization, with the ambition to extract common tasks from electronic structure programs and redesign them as free, open-source libraries. They include “heavy-duty” ones with a high degree of parallelisation, and potential for adaptation to novel hardware within them, thereby separating the sophisticated computer science aspects of performance optimization and re-engineering from the computational science done by scientists when implementing new ideas. It is a community effort, undertaken by developers of various successful codes, now facing the challenges arising in the new model. This modular paradigm will improve overall coding efficiency and enable specialists (computer scientists or computational scientists) to use their skills more effectively. It will lead to a more sustainable and dynamic evolution of software as well as lower barriers to entry for new developers.
Maximally-localised Wannier functions (MLWFs) are routinely used to compute from first- principles advanced materials properties that require very dense Brillouin zone (BZ) integration and to build accurate tight-binding models for scale-bridging simulations. At the same time, high-thoughput (HT) computational materials design is an emergent field that promises to accelerate the reliable and cost-effective design and optimisation of new materials with target properties. The use of MLWFs in HT workflows has been hampered by the fact that generating MLWFs automatically and robustly without any user intervention and for arbitrary materials is, in general, very challenging. We address this problem directly by proposing a procedure for automatically generating MLWFs for HT frameworks. Our approach is based on the selected columns of the density matrix method (SCDM, see SCDM Wannier Functions) and is implemented in an AiiDA workflow.
Purpose of the module
Create a fully-automated protocol based on the SCDM algorithm for the construction of MLWFs, in which the two free parameters are determined automatically (in our HT approach the dimensionality of the disentangled space is fixed by the total number of states used to generate the pseudopotentials in the DFT calculations).
A paper describing the work is available at https://arxiv.org/abs/1909.00433, where this approach was applied to a dataset of 200 bulk crystalline materials that span a wide structural and chemical space.
Background information
This module is a collaboration between E-CAM and the MaX Centre of Excellence.
In the SCDM Wannier Functions module, E-CAM has implemented the SCDM algorithm in the pw2wannier90.f90 interface code between the Quantum ESPRESSO software and the Wannier90 code. This implementation was used as the basis for a complete computational workflow for obtaining MLWFs and electronic properties based on Wannier interpolation of the BZ, starting only from the specification of the initial crystal structure. The workflow was implemented within the AiiDA materials informatics platform, and used to perform a HT study on a dataset of 200 materials, as described in here.
Quantum Monte Carlo (QMC) methods are a class of ab initio, stochastic techniques for the study of quantum systems. While QMC simulations are computationally expensive, they have the advantage of being accurate, fully ab initio and scalable to a large number of cores with limited memory requirements.
These features make QMC methods a valuable tool to assess the accuracy of DFT computations, which are widely used in the fields of condensed matter physics, quantum chemistry and material science.
QMCPack is a free package for QMC simulations of electronic structure developed in several national labs in the US. This package is written in object oriented C++, offers a great flexibility in the choice of systems, trial wave functions and QMC methods and supports massive parallelism and the usage of GPUs.
Trial wave functions for electronic QMC computations commonly require the use of single electrons orbitals, typically computed by DFT. The aim of the E-CAM pilot project described here is to build interfaces between QMCPack and other softwares for electronic structure computations, e.g. the DFT code Quantum Espresso.
These interfaces are used to manage the orbital reading or their DFT generation within QMCPack, to establish an automated, black box workflow for QMC computations. QMC simulation can for example be used in the benchmark and validation of DFT calculations: such a procedure can be employed in the study of several physical systems of interest in condensed matter physics, chemistry or material science, with application in the industry, e.g. in the study of metal-ion or water-carbon interfaces.
The following modules have been built as part of this pilot project:
QMCQEPack, that provides the files to download and properly patch Quantum Espresso 5.3 to build the libpwinterface.so library; this library is required to use the module ESPWSCFInterface to generate single particle orbitals during a QMCPack computation using Quantum Espresso.
ESInterfaceBase that provides a base
class for a general interface to generate single particle orbitals to be
used in QMC simulations in QMCPack; implementations of specific interfaces as derived
classes of ESInterfaceBase are available as the separate modules as follows:
PANNA is a package for training and validating neural networks to
represent atomic potentials. It implements configurable all-to-all connected
deep neural network architectures which allow for the exploration of training
dynamics. Currently it includes tools to enable original[1] and modified[2]
Behler-Parrinello input feature vectors, both for molecules and crystals, but
the network can also be used in an input-agnostic fashion to enable further
experimentation. PANNA is written in Python and relies on TensorFlow as
underlying engine.
A common way to use PANNA in its current implementation is to train a
neural network in order to estimate the total energy of a molecule or crystal,
as a sum of atomic contributions, by learning from the data of reference total
energy calculations for similar structures (usually ab-initio calculations).
The neural network models in literature often start from a description
of the system of interest in terms of local feature vectors for each atom in
the configuration. PANNA provides tools to calculate two versions of the
Behler-Parrinello local descriptors but it allows the use of any
species-resolved, fixed-size array that describes the input data.
PANNA allows the construction of neural network architectures with
different sizes for each of the atomic species in the training set. Currently the
allowed architecture is a deep neural network of fully connected layers,
starting from the input feature vector and going through one or more hidden
layers. The user can determine to train or freeze any layer, s/he can also
transfer network parameters between species upon restart.
In summary, PANNA is an easy-to-use interface for obtaining neural network models for atomistic potentials, leveraging the highly optimized TensorFlow infrastructure to provide an efficient and parallelized, GPU-accelerated training.
It provides:
an input creation
tool (atomistic calculation result -> G-vector )
an input packaging
tool for quick processing of TensorFlow ( G-vector -> TFData bundle)
[1] J. Behler and M. Parrinello, “Generalized Neural-Network Representation
of High-Dimensional Potential-Energy Surfaces”,
Phys. Rev. Lett. 98, 146401 (2007)
[2] Justin S. Smith, Olexandr Isayev, Adrian E. Roitberg, “ANI-1: An
extensible neural network potential with DFT accuracy at force field
computational cost», Chemical Science,(2017), DOI: 10.1039/C6SC05720A
[3] Justin S. Smith, Olexandr Isayev, Adrian E. Roitberg, “ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules; Scientific Data, 4 (2017), Article number: 170193, DOI: 10.1038/sdata.2017.193
FFTXlib is mainly a rewrite and optimisation of earlier versions of FFT related routines inside Quantum ESPRESSO (QE) pre-v6; and finally their replacement. Despite many similarities, current version of FFTXlib dramatically changes the FFT strategy in the parallel execution, from 1D+2D FFT performed in QE pre v6 to a 1D+1D+1D one; to allow for greater flexibility in parallelisation.
Practical application and exploitation of the code
FFTXlib module is a collection of driver routines that allows the user to perform complex 3D fast Fourier transform (FFT) in the context of plane wave based electronic structure software. It contains routines to initialize the array structures, to calculate the desired grid shapes. It imposes underlying size assumptions and provides correspondence maps for indices between the two transform domains.
Once this data structure is constructed, forward or inverse in-place FFT can be performed. For this purpose FFTXlib can either use a local copy of an earlier version of FFTW (a commonly used open source FFT library), or it can also serve as a wrapper to external FFT libraries via conditional compilation using pre-processor directives. It supports both MPI and OpenMP parallelisation technologies.
FFTXlib is currently employed within Quantum Espresso package, a widely used suite of codes for electronic structure calculations and materials modeling in the nanoscale, based on planewave and pseudopotentials.
FFTXlib is also interfaced with “miniPWPP” module that solves the Kohn Sham equations in the basis of planewaves and soon to be released as a part of E-CAM Electronic Structure Library.
Software documentation and link to the source code can be found in our E-CAM software Library here.
MatrixSwitch is a module which acts as an intermediary interface layer between high-level and low-level routines dealing with matrix storage and manipulation. It allows a seamlessly switch between different software implementations of the matrix operations.
DBCSR is an optimized library to deal with sparse matrices, which appear frequently in many kind of numerical simulations.
In DBCSR@MatrixSwitch, DBCSR capabilities have been added to MatrixSwitch as an optional library dependency.
To carry out calculations in serial mode may be too slow sometimes and a parallelisation strategy is needed. Serial/parallel MatrixSwitch employs Lapack/ScaLapack to perform matrix operations, irrespective of their dense or sparse character. The disadvantage of the Lapack/ScaLapack schemes is that they are not optimised for sparse matrices. DBCSR provides the necessary algorithms to solve this problem and in addition is specially suited to work in parallel.
The State-of-the-Art workshop in the E-CAM Electronic Structure Work-Package (WP2) gathered together 38 participants from the academic research world, shared in a rather equilibrated fashion among Density Functional Theory, Quantum Monte Carlo and Machine Learning communities, and one industrial researcher from Scienomics. Key topics to the development of the field of computational materials science from first principles were thoroughly discussed, from which the following outcomes have emerged: (1) Importance of computational benchmarks to assess the accuracy of different methods and to feed the machine learning and neural network schemes with reliable data; (2) Need of a common database, and need to develop a common language across different codes and different computational approaches; (3) Interesting capabilities for neural network methods to develop new correlated wave functions; (4) Cross-fertilizing combination of computational schemes in a multi-scale environment; and (5) Recent progress in Quantum Monte Carlo to further improve the accuracy of the calculations by taking alternative routes. Limitations in the field and open questions were also debated, as described in the workshop scientific report.