This work relates to the implementation of a performance portable version of DL_MESO (DPD) using the Kokkos library. It focuses on porting to DL_MESO (DPD) the first and second loops of the Verlet Velocity (VV) scheme for the time marching scheme. This allows to run DL_MESO on NVidia GPUs as well as on other GPUs or architectures (many-core hardware like KNL), allowing performance portability as well as separation of concern between computational science and HPC.
Description
The VV scheme is made of 3 steps:
a first velocity and particle positions integration by Delta t/2,
a force calculation, and
a second velocity integration by Delta t/2.
Steps 1) and 2) are documented is the following two modules
Note: Kokkos is a C++ library, while DL_MESO (DPD) is in Fortran90 Language. The current implementation requires a transfer between Fortran to C++, due to the use of Fortran pointers not bound using the ISO_C_BINDING standard. This constraint will be removed in future versions of DL_MESO.
Practical application
With the advent of heterogeneous hardware, achieving performance portability across different architectures is one of the main challenges in HPC. In fact, while specific languages, like CUDA, can give best performance for the NVidia hardware, they cannot be used with different GPU vendors limiting the usage across supercomputers worldwide.
In this module we use Kokkos, developed at Sandia National Laboratories, which consist of several C++ templated libraries which provide the capability to offload a workload to several different architectures, taking care of the memory layout and transfer between host and device.
Documentation and source code
The modules documentation is available on our software repository here. The modules have also been pushed into DL_MESO git repository as explained in the modules documentation.
This module describes the work done in E-CAM in cooperation with the HemeLB code from the CompBioMed Centre of Excellence.
HemeLB is a high performance lattice-Boltzmann solver optimised for simulating blood flow through sparse geometries, such as those found in the human vasculature. The code is used within the CompBioMed HPC Centre of Excellence H2020 project and is already highly optimised for HPC usage. Nevertheless, in an E-CAM workshop on the load balancing library ALL hosted at the Juelich Supercomputing Centre, a cooperation was set up in order to analyse and test whether the use of ALL could improve the existing scalability of the code.
ALL was designed to work with particle codes, therefore it was interesting to apply the library to a lattice-Boltzmann solver, which usually is not particle-based. The different grid points of the solution grid were designated as particles and since each of the grid-points already was assigned a workload, the sum of grid-point workloads could be used as domain work load.
As a result, it was demonstrated that the domain compositions provided by ALL show a better theoretical load distribution. Tests to check if this translates into better code performance are inconclusive as yet, due to hardware related issues on the testing platforms. However, these are currently under further investigation, and more definitive results about the performance of the ALL-provided domain decompositions can be expected in the near future. The results were part of an article about HemeLB, which was published in 2020[1] .
Towards blood flow in the virtual human: efficient self-coupling of HemeLB J. W. S. McCullough, R. A. Richardson, A. Patronis, R. Halver, R. Marshall, M. Ruefenacht, B. J. N. Wylie, T. Odaker, M. Wiedemann, B. Lloyd, E. Neufeld, G. Sutmann, A. Skjellum, D. Kranzlmüller and P. V. Coveney Interface Focus2020, 11: 20190119 DOI: http://dx.doi.org/10.1098/rsfs.2019.0119 (open access)
To further expand the portfolio of activities targeted at industrialists, E-CAM has established a series of new events targeted at training interested industrial researchers on the simulation and modelling techniques implemented in specific codes and in the direct use of this software for their industrial applications.
The first event of this series will focus on the area of meso- and multiscale simulations and on the flagship code DL_MESO:
In this workshop we will introduce DL_MESO: a software package for mesoscale simulations. Usage of the software will be gradually presented, starting with tutorials based on theoretical background and following up with hands-on sessions. We will focus on the Dissipative Particle Dynamics (DPD) methodology, exploring the different capabilities of DL_MESO_DPD via practical examples that reflect daily industrial challenges.
DL_MESO has been used for a wide range of problems of both scientific and industrial interest. The code is used, for example, in projects with Unilever, Syngenta and Infineum – to develop DPD parameterisation strategies and simulation protocols to predict important properties of newly-devised surfactant-based formulations; with IBM Research Europe – to model nanofluidic multiphase. The code developers themselves will provide the training. The event is co-organized by Formeric, a company that helps industrial users to study their own formulated projects, primarily by developing a software platform to make it easier for them to access DPD simulations and modelling tools.
As part of the event, UKRI STFC offers a 6-month one seat free licence of DL_MESO 2.7 to be used soon after the end of the event, which will help testing the software.
Don’t miss this opportunity to be trained by the experts on the methods and on the codes themselves! Register for event at
Our last Extended Software Development Workshop (ESDW) took place on the 18th-22nd January[1], and given its length (5 days) and it’s nature (theory and hands-on training sessions) it was a real success! “The workshop went very well, participants seem to have enjoyed and they lasted until the end !”, said organiser Jony Castagna, computational scientist and E-CAM programmer at UKRI STFC Daresbury Laboratory. The event, organised at the CECAM-UK-DARESBURY Node[2], focused on HPC for mesoscale simulation, and aimed at introducing participants to Dissipative Particle Dynamics (DPD) and the mesoscale simulation package DL_MESO [3] (DL_MESO_DPD). DL_MESO is developed at UKRI STFC Daresbury by Michael Seaton, computational chemist at Daresbury and also an organiser of this event.
Another component of this workshop was parallel programming of hybrid CPU-GPU systems. In particular, DL_MESO has recently been ported to multi-GPU architectures[4] and runs efficiently up to 4096 GPUs, an effort supported by E-CAM (thank you Jony!). Part of this workshop was dedicated to theory lectures and hands-on sessions on GPU architectures and OpenACC (NVidia DLI course) given by Jony, which is an NVidia DLI Certified Instructor. He said “The intention is not only to port mesoscale solvers on GPUs, but also to expose the community to this new programming paradigm, which they can benefit from in their own fields of research”.
All sessions in this ESDW were followed by discussions and hands-on exercises. Organisers were supported by another STFC colleague and former E-CAM post-doc Silvia Chiacchiera. One of the participants wrote “Thank you so much for your effort. This workshop will cause a significant shift in my thinking and approach”.
21 people registered for to the event; but by the third day there were only 9… from which 5 lasted until the last session! A picture taken from the last session talks by itself 🙂
Do you want to join our next training event ? Check out our programme :
Scalability of parallel applications depends on a number of characteristics, among which is efficient communication, equal distribution of work or efficient data lay-out. Especially for methods based on domain decomposition, as it is standard for, e.g., molecular dynamics, dissipative particle dynamics or particle-in-cell methods, unequal load is to be expected for cases where particles are not distributed homogeneously, different costs of interaction calculations are present or heterogeneous architectures are invoked, to name a few. For these scenarios the code has to decide how to redistribute the work among processes according to a work sharing protocol or to dynamically adjust computational domains, to balance the workload. The A Load Balancing Library (ALL) developed within E-CAM at the Julich Supercomputing Center aims to provide an easy and portable way to include dynamic domain-based load balancing into particle based simulation codes. It provides several schemes to find the ideal split of the workload, from the simplest orthogonal non staggered domain decomposition, to the more fancy Voronoi mesh scheme. Within this text we provide an overview of ALL, its capabilities and current use cases, as well as where to find additional information on the library.
Most modern parallelized (classical) particle simulation programs are based on a spatial decomposition method as an underlying parallel algorithm: different processors administrate different spatial regions of the simulation domain and keep track of those particles that are located in their respective region. Processors exchange information
in order to compute interactions between particles located on different processors
to exchange particles that have moved to a region administered by a different processor.
This implies that the workload of a given processor is very much determined by its number of particles, or, more precisely, by the number of interactions that are to be evaluated within its spatial region.
Certain systems of high physical and practical interest (e.g. condensing fluids) dynamically develop into a state where the distribution of particles becomes spatially inhomogeneous. Unless special care is being taken, this results in a substantially inhomogeneous distribution of the processors’ workload. Since the work usually has to be synchronized between the processors, the runtime is determined by the slowest processor (i.e. the one with the highest workload). In the extreme case, this means that a large fraction of the processors are idle during these waiting times. This problem becomes particularly severe if one aims at strong scaling, where the number of processors is increased at constant problem size: Every processor administrates smaller and smaller regions and therefore inhomogeneities will become more and more pronounced. This will eventually saturate the scalability of a given problem, already at a processor number that is still so small that communication overhead remains negligible.
The solution to this problem is the inclusion of dynamic load balancing techniques. These methods redistribute the workload among the processors, by lowering the load of the most busy cores and enhancing the load of the most idle ones. Fortunately, several successful techniques are known already to put this strategy into practice. Nevertheless, dynamic load balancing that is both efficient and widely applicable implies highly non-trivial coding work. Therefore it has not yet been implemented in a number of important codes.
The A Load-Balancing Library (ALL) developed within E-CAM at the Simulation Laboratory Molecular Systems of the Juelich Supercomputing Centre, aims to provide an easy and portable way to include dynamic domain-based load balancing into particle based simulation codes. It was created in the context of an Extended Software Development Workshop (ESDW) within E-CAM (see ALL ESDW event details), where code developers of CECAM community codes were invited together with E-CAM postdocs, to work on the implementation of load balancing strategies. The goal of this activity is to increase the scalability of applications to a larger number of cores on HPC systems, for spatially inhomogeneous systems, and thus to reduce the time-to-solution of the applications .
Particle system before and after the load balancing. Left: equal domain sizes with bad balance; right: unequal domain sizes and good work load.
ALL includes several load-balancing schemes, with additional approaches currently being added. The following list gives an overview about the currently included schemes:
Tensor-Product method: For the Tensor-Product method, the work on all processes (subdomains) is reduced over the cartesian planes in the systems. This work is then equalized by adjusting the borders of the cartesian planes.
Staggered Grid Method: For the staggered-grid scheme, a 3-step hierarchical approach is applied: work over the Cartesian planes is reduced before the borders of these planes are adjusted; in each of the Cartesian planes the work is reduced for each Cartesian column, these columns are then adjusted to each other to homogenise the work in each column; the work between neighbouring domains in each column is adjusted. Each adjustment is done locally with the neighbouring planes, columns or domains by adjusting the adjacent boundaries.
Unstructured Mesh Method: In contrast to the Tensor-Product method and the Staggered Grid Method, the unstructured mesh method adjusts domains not by moving boundaries but vertices, i.e. corner points, of domains. For each vertex, a force, based on the differences in work of the neighboring domains, is computed and the vertex is shifted in a way to equalize the work between these neighboring domains.
Voronoi Mesh Method: Similar to the topological mesh method (Unstructured Mesh Method), the Voronoi mesh method computes a force, based on work differences. In contrast to the topological mesh method, the force acts on a Voronoi point rather than a vertex, i.e. a point defining a Voronoi cell, which describes the domain. Consequently, the number of neighbors is not a conserved quantity, i.e. the topology may change over time.
Histogram-based Staggered Grid Method: The histogram-based staggered-grid scheme results in the same grid as the staggered-grid scheme (see Staggered Grid Method), this scheme uses the cumulative work function in each of the three cartesian directions in order to generate this grid. Using histograms and the previously defined distribution of process domains in a cartesian grid, this scheme generates in three steps a staggered-grid result, in which the work is distributed as evenly as the resolution of the underlying histogram allows. In contrast to the other schemes this scheme depends on a global exchange of work between processes.
Use cases
ALL is being tested with the HemeLB code[1] from the Centre of Excellence CompBiomed. A recent paper describes how HemeLB’s developments in memory management and load balancing (with ALL) allow near linear scaling performance of the code on hundreds of thousands of computer codes[2].
ALL is implemented in the multi-GPU version of DL_MESO_DPD package (see related news item here). The intention of this integration is to allow for better performance when modelling complex systems with DL_MESO_DPD[3], like large proteins or lipid bilayers, redistributing the work load across the GPUs.
References
[1] D. Groen, J. Hetherington, H.B. Carver, R.W. Nash, M.O. Bernabeu, and P.V. Coveney. Analysing and modelling the performance of the HemeLB lattice-Boltzmann simulation environment. Journal of Computational Science, 4(5):412 – 422, 2013. doi: https://doi.org/10.1016/j.jocs.2013.03.002. // HemeLB URL: www.hemelb.org
[2] McCullough JWS et al. 2021 Towards blood flow in the virtual human: efficient self-coupling of HemeLB. Interface Focus 11: 20190119. doi: http://dx.doi.org/10.1098/rsfs.2019.0119
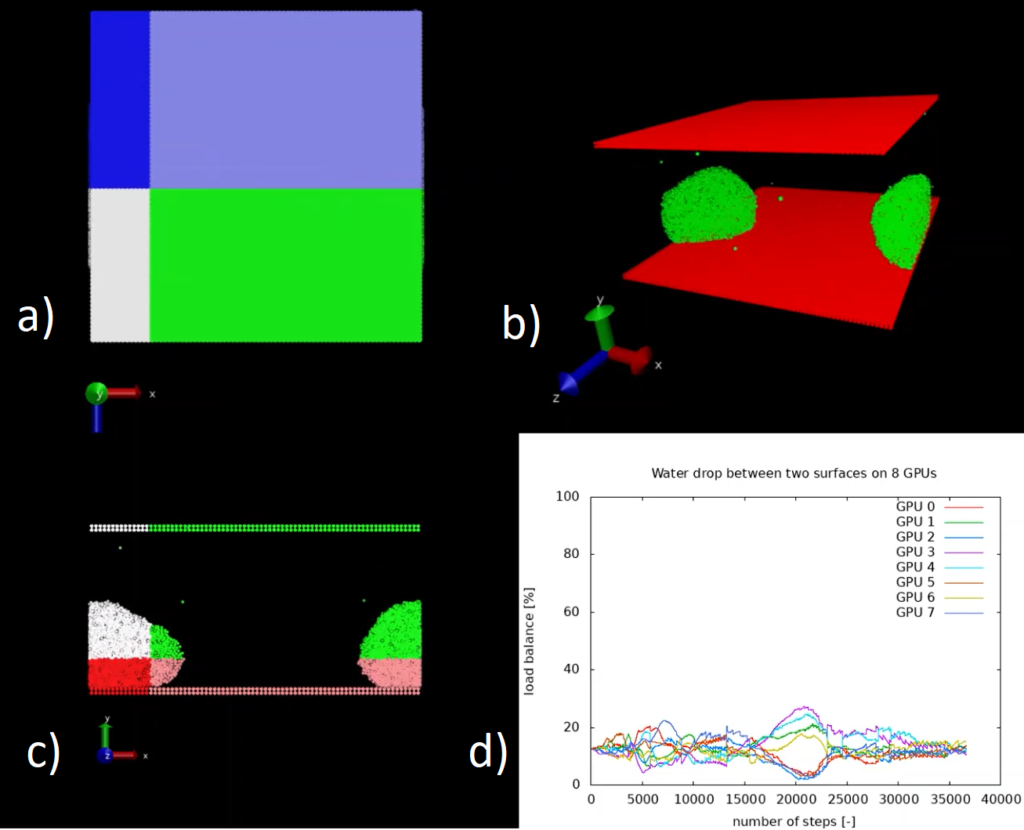
A test case was implemented (see Figure 1 a), b) and c)) that reproduces 32k water beads initially scattered along a regular structure and then slowly agglomerating towards an unique large drop confined between two parallel surfaces. The system is divided across 8 GPUs and, for the purposes of the visualisation, we restrict ourselves to 32k particles. For a larger number of particles it would not be possible to simulate the system without load-balancing, since all the particles agglomerate to a subset of the available GPUs and one or more GPUs would run out of memory having to accommodate a large number of particles. Moreover, such a strong load imbalance drastically reduces the scalability of the application.
In Figure d) we see the time history of the load imbalance for each GPU when using the ALL library. Without load balancing the system would gradually diverge from the ideal value of 12.5%. You can find a video that shows the evolution of the load-balancing for this system in another software module.
Figure 1: Load imbalance in DL_MESO with ALL library for a water drop between two surfaces. Each colour represents different domain assigned to a different GPU: a) top view, b) perspective view, c) front view, d) load balance vs time
Source code
Further details on the implementation of ALL library in DL_MESO and the source code can be found in the E-CAM software repository here.
Few software, like DL_MESO, userMESO and LAMMPS, can currently simulate large Dissipative Particle Dynamics (DPD) simulations. In particular, DL_MESO [1, 2] has recently been ported to multi-GPU architectures and runs efficiently up to 4096 GPUs, an effort supported by E-CAM.
In this E-CAM Extended Software Development Workshop, the developers of the DL_MESO code themselves will provide an introduction to DPD, DL_MESO, its features and functionalities, as well as they will initiate participants to parallel programming of hybrid CPU-GPU systems. Part of the workshop will be dedicated to theory lectures and hands-on sessions on GPU architectures and OpenACC (NVidia DLI course) given by an NVidia DLI Certified Instructor, followed by the practical case of porting DL_MESO to OpenACC.
Interested in participating? Join us on the 18-22 January for this ONLINE course. Express your motivation to attend the workshop directly through the CECAM website at https://www.cecam.org/workshop-details/8
[2] M. A. Seaton, R. L. Anderson, S.Metz, and W. Smith, “DL_MESO: highly scalable mesoscale simulations,”Molecular Simulation, vol. 39, no. 10, pp. 796–821, Sep. 2013
A community-driven review with contributions from E-CAM “Unfolding the prospects of computational (bio)materials modeling”has just been published in the Journal of Chemical Physics on the history, developments, and challenges facing coarse graining (CG) and multiscale simulation (MS) and a set of recommendations on how the latter may be addressed.
E-CAM researchers working at the Hartree Centre – Daresbury Laboratory have co-designed the DL_MESO Mesoscale Simulation package to run on multiple GPUs, and ran for the first time a Dissipative Particle Dynamics simulation of a very large system (1.8 billion particles) on 4096 GPUs.
Towards extreme scale dissipative particle dynamics simulations using multiple GPGPUs J. Castagna, X. Guo, M. Seaton and A. O’Cais Computer Physics Communications (2020) 107159 DOI: 10.1016/j.cpc.2020.107159 (open access)
Abstract
A multi-GPGPU development for Mesoscale Simulations using the Dissipative Particle Dynamics method is presented. This distributed GPU acceleration development is an extension of the DL_MESO package to MPI+CUDA in order to exploit the computational power of the latest NVIDIA cards on hybrid CPU–GPU architectures. Details about the extensively applicable algorithm implementation and memory coalescing data structures are presented. The key algorithms’ optimizations for the nearest-neighbour list searching of particle pairs for short range forces, exchange of data and overlapping between computation and communications are also given. We have have carried out strong and weak scaling performance analyses with up to 4096 GPUs. A two phase mixture separation test case with 1.8 billion particles has been run on the Piz Daint supercomputer from the Swiss National Supercomputer Center. With CUDA aware MPI, proper GPU affinity, communication and computation overlap optimizations for multi-GPU version, the final optimization results demonstrated more than 94% efficiency for weak scaling and more than 80% efficiency for strong scaling. As far as we know, this is the first report in the literature of DPD simulations being run on this large number of GPUs. The remaining challenges and future work are also discussed at the end of the paper.
GC-AdResS is a technique that speeds up computations without loss of accuracy for key system properties by dividing the simulation box into two or more regions having different levels of resolution, for instance a high resolution region where the molecules of the system are treated at an atomistic level of detail, and other regions where molecules are treated at a coarse grained level, and transition regions where a weighted average of the two resolutions is used. The goal of the E-CAM GC-AdResS pilot project was to eliminate the need of a transition region so as to significantly improve performance, and to allow much greater flexibility. For example, the low resolution region can be a particle reservoir (ranging in detail from coarse grained to ideal gas particles) and a high resolution atomistic region with no transition region, as was needed hitherto. The only requirement is that the two regions can exchange particles, and that a corresponding “thermodynamic” force is computed self-consistently, which it turns out is very simple to implement.