Challenges to Industry of drug substance development
Computation based methods play a growing role in all stages of accelerated medicine pipelines responding to industry challenges of drug substance development.
Abstract
APC was created In 2011 by Dr. Mark Barrett and Prof. Brian Glennon of the University College Dublin School of Chemical and Bioprocess Engineering with a mission to harness state of the art research methods & know-how to accelerate drug process development. Since then it has grown organically partnering with companies across the world, large and small, to bring medicines to market at unprecedented speed. Computation-based methods play a growing role in all stages of its medicine pipeline as explained by Dr. Jacek Zeglinski in this E-CAM interview on the challenges to Industry of drug substance development.
What is APC?
APC, the company I work for stands for Applied Process Company, and we are based in South County Dublin in Ireland, actually, just half an hour walk from the Irish Sea. APC is a global company, although it’s perhaps not very big, it is an impactful company because we collaborate with a range of big pharma companies across the world, both big and small. We provide services to them to help accelerate the development of active pharmaceutical ingredients (APIs), both small molecules, and biomolecules. We work in a number of domains of product development, starting from early-stage development through to optimization up to scale up and technology transfer.
Can you tell us how you got involved with APC?
I learned about APC in 2012, while working as a postdoc at the University of Limerick. At that time APC was a newly established start up company. Some time later two friends from my research group joined APC, so very soon I had first-hand opinion of the company. It appeared that the superior company culture and the dive-deep research focus were sufficiently strong arguments for me to join APC in early 2019.
What challenges does APC focus on, say in the context of drug development?
When we refer to drugs or drug substances, we have in mind the powder forms that are the basis for making tablets. Usually, they are crystalline but sometimes they are in amorphous form. There are a number of challenges we face. The poor solubility of active ingredients is probably the most important. Until recently people would have considered APIs to be small molecules, but nowadays they are getting larger and larger, up to 1000 grams per mole in molecular weight. They are also quite flexible and complex and that actually brings challenges related to their solubility. Most of the APIs we are handling have poor solubility in a range of solvents. That would translate to insufficient bio-availability, and make it difficult to manufacture those APIs with high productivity (i.e. producing low yield and throughput) if we could not solve the problem of solubility.
The second challenge we face is polymorphism. Crystalline materials can exist in a number of different crystal forms, those forms are interrelated in terms of stability, some of them are metastable and the most stable ones and not always easy to obtain. Sometimes one gets unwanted solvates or hydrates at the start of the development process when screening is being done in a range of different solvents. The third issue relates to particle size and shape, which can cause poor processability, filter-ability, flow-ability, compressibility, and difficulty to form tablets. Particles that are too large can have poor or inconsistent dissolution rates and bioavailability. Agglomeration is another challenge, and consequent difficulties to remove impurities, particularly when they are hidden in the voids within agglomerates.
What computational approaches do you use?
In our work in optimizing processes, we mainly do a lot of experiments but we also use computational methods. I will briefly highlight the latter, starting with our typical workflow for solvent selection for crystallization processing.
Solubility Predictions for Crystallisation Processes
Computational tools allow us to screen many more solvents than would be possible using experimentation alone. The workflow usually starts with 70 solvents, which are screened computationally regarding their solubility temperature dependence, the propensity of forming solvates or hydrates, and their utility for impurity purging, ending up with less than 10 promising solvents for experimental validation. This computational screening which is done in two to three days would take several weeks or even months were experiment used instead. We then check for the crystal form, for purity, and some other parameters that allow us to narrow down our candidates to two, three, or even one solvent for small-scale crystallisation experiments. The most optimum solvent system is further used to develop the full process followed by a final scale-up demonstration. So as you can see that computational part is a very important piece of this workflow and it really aids the solvent selection process. Regarding software, we are using mainly two platforms from Biovia, one is COSMO-therm (combined with a DFT-based program – TURBOMOLE) and the other one is Materials Studio. With COSMO we predict solubilities, and also the propensity for solvate/cocrystal and even salt formation. With Materials Studio, we search for the low energy conformations of molecules and use those conformations for solubility prediction using COSMO, so the two software codes work together. With Materials Studio, we also do lattice and cohesive energy calculations, so as to estimate thermodynamic stability of polymorphs, or find imperfections in the crystal lattice or predict the shape of crystals. We are constantly developing these capabilities and trying to do more predictive modeling. Looking at the molecular complexity of the future medicines we handle, we clearly see that there is a real need to better understand the solid-state features of those APIs, which very often are multicomponent materials, e.g. cocrystals, salt-cocrystal-hydrate hybrids etc.
How do the computational solubility predictions compare with experiment?
I would like to show how relative solubility predictions performed with COSMO compare with experiments for a medium-sized API molecule (500g/Mol) in a variety of solvents. As you can see there’s a good match between the rank order of solubility.
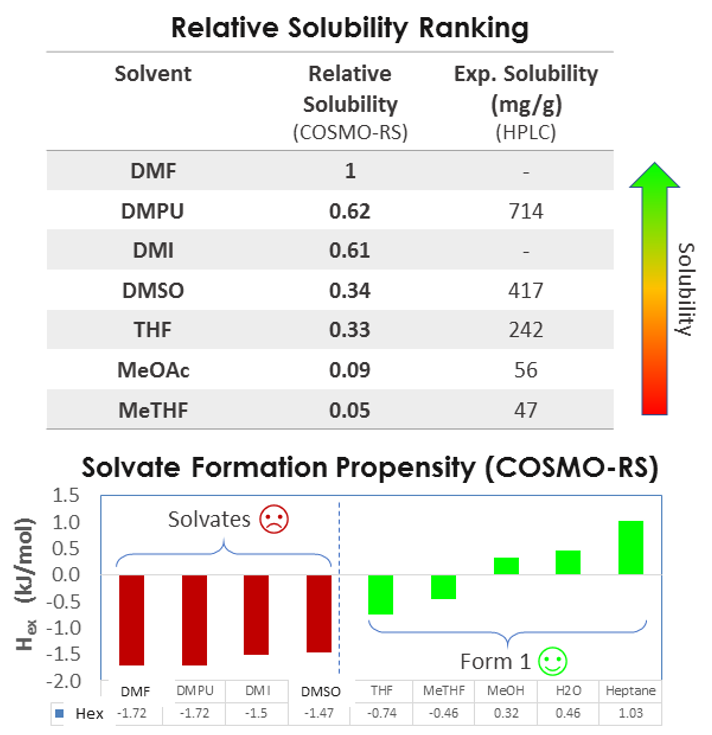
So we can predict relative solubility and, in addition, we can compute the propensity for solvate formation, that is the enthalpy of mixing, which is related to the interaction between solvent and solute. More negative values indicate exothermic events and stronger interactions, and more positive values indicate endothermic events and unfavorable mixing. So the case study presented was a very successful validation of this approach. In addition to relative solubility, we can predict absolute solubility and compare it with experiment, for example at two different temperatures, 20 and 48 °C.
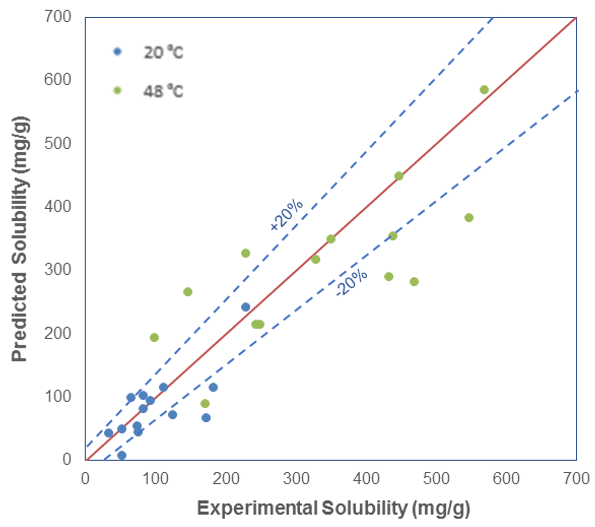
As you can see, for some of the systems, the solubility is predicted very well, but for most systems, the computational and experimental results are within 20% of each other. At lower temperatures, the agreement is very good, but at higher temperatures, we see the predictions are not as accurate.
In addition to single solvents, we can predict solubility in solvent mixtures. This is a very useful application because, for many APIs, there is no single neat solvent that can be identified. In such cases, there is often a solvent mixture that can be identified which gives rise to good solubility for the API. For example, in the binary solvent mixture of water and ethyl acetate, certain solvent ratios give rise to good solubility for a variety of solutes. We can also have ternary, quaternary, and higher mixtures of solvents studied computationally.
How important is the stability of different conformers of an API in a solvent?
Conformational Aspects in Solubility Predictions
This is perhaps more fundamental, and interesting. I will show an example of solubility predictions for malic acid, and focus on different conformations. Malic acid can make a number of intra-molecular hydrogen bonds and that will be reflected in different relative conformational energies. There’s a hypothesis that regarding solubility predictions, the lowest energy conformations should be dominant because this is the confirmation that should be the most probable and most frequently occurring in solution. However, we find that this is not always the case. We can compare the predicted values of solubilities for five different solvents with experimental values for the crystal-like molecular conformation obtained in vacuum with DFT using COSMO and ten other low energy conformers identified based on their relative energetic stability.
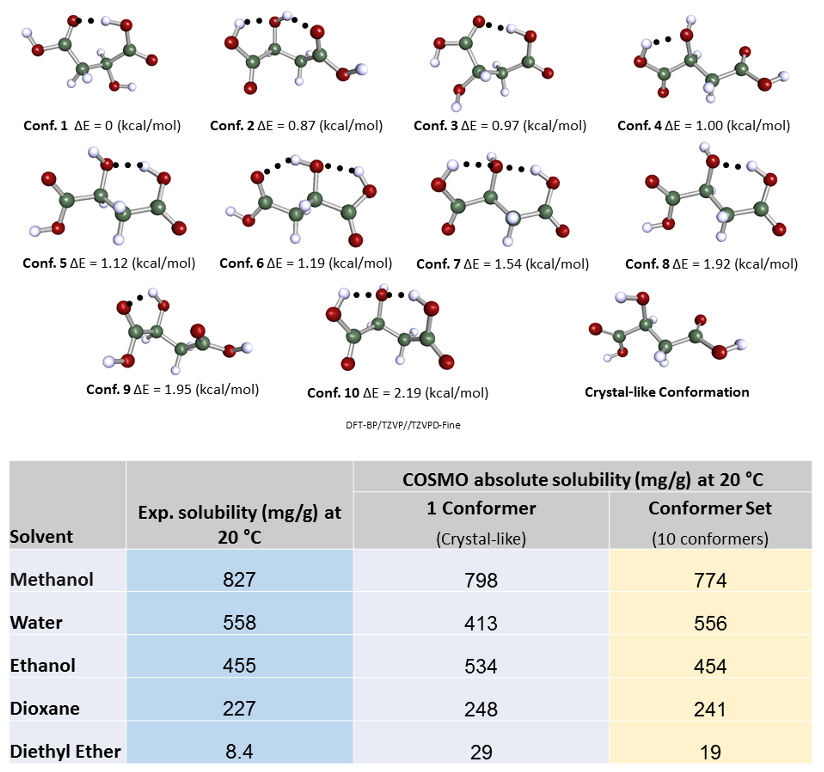
As we can see the predicted solubility compare fairly well with experiment, particularly in terms of relative ranking of solubility, as this allows the best solvent to be chosen. We see that the comparison using ten conformers is slightly better for predicting absolute solubility. However, it is not always the case that we have such good agreement for solubilities between prediction and experiment. For molecules that are larger and more flexible, the challenge increases. Another case we have studied is tolbutamide. In polar solvents such as methanol, the molecule should not have an intra-molecular hydrogen bond conformation, but in non-polar or low polarity solvents such as toluene, it should. We generated a variety of conformations some of which had intra-molecular hydrogen bonds and predicted the solubilities using those different conformations having different relative energies.
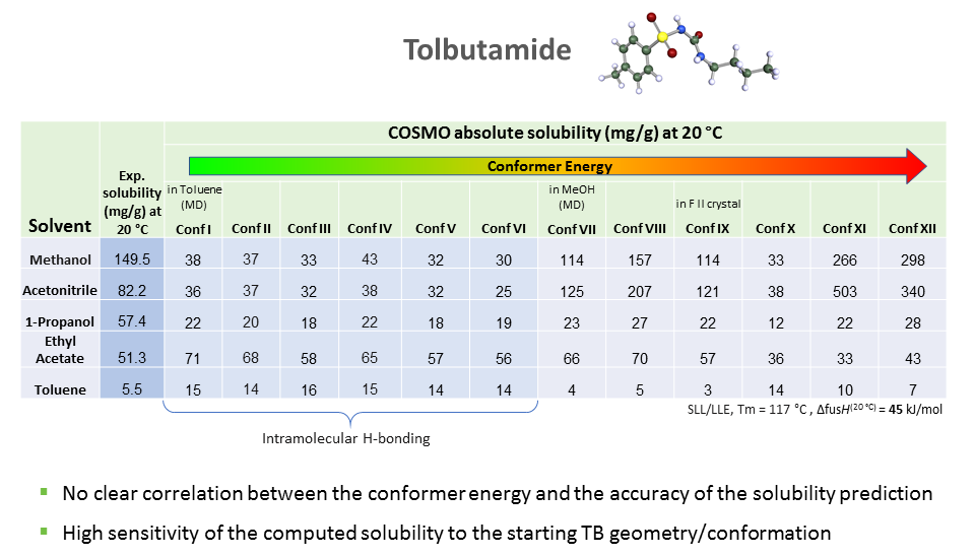
The hypothesis that the lower the energy of the conformer, the better its solubility does not apply in this case. There is no clear correlation between conformer energy and the accuracy of the solubility predicted when compared with the experiment for tolbutamide.
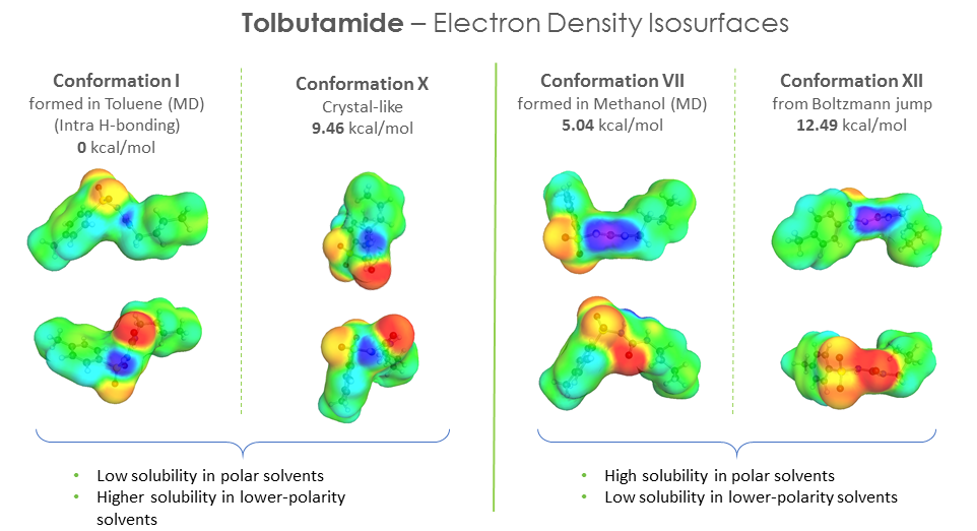
We also looked at the electron density iso-surfaces for the different conformers and found a correlation between the polar “domains” in the distribution (local concentration of positive and negative charge distributions) and the solubility in polar solvents and vice-versa for non-polar solvents – where the distribution of positive and negative charges were more scattered and less pronounced. It would be nice to quantify this and explore it further.
How do you predict the morphology of APIs and how well do they compare with experiments?
Crystal Shape/Morphology Prediction of Pharmaceutical Compounds
In our projects, before starting any experiments we try to predict as many properties as possible, including shape/morphology of crystals of a particular API. The predictions are made in vacuum, and very often reflect the experimental morphology. There are some exceptions, but in most cases, we find good agreement. We use an attachment energy theory which assumes that crystal growth is proportional to the attachment energy, i.e. the energy released when a slice of crystal is added to a particular facet (or lattice direction). The higher the attachment energy on the facet, the faster the growth in that direction. Based on that, we can predict the shape of the crystalline particle and the intrinsic propensity for its growth in particular directions, which will also give us the theoretical aspect ratio. It turns out that this is often related to the relative polarity of different facets; basically the number of polar atoms at different surfaces. Usually, the higher the polarity, the stronger the hydrogen bonding, and the faster will be the growth rate in that direction. However, there can be exceptions to that. For example, for molecules that are elongated and somewhat flat like Clofazimine, there are no intermolecular hydrogen bonds that occur, and molecular packing is more important. Ideally, we would like to predict the effect of the solvent on the crystal shape. Sometimes we are able to find a correlation between the surface chemistry and the morphology we are trying to predict and the impact of a particular solvent type. For example, some systems have high polarity charge distributions on certain facets, and in such cases, if we use polar-protic solvents, they would preferentially interact with polar atoms on that facet and inhibit crystal growth there. On the other hand, they could allow growth along low polarity directions. We found some experimental preliminary evidence of this, but it still has to be confirmed. We are trying to use this sort of analysis to predict which solvent will generate the desired morphology. We also explore more complex modelling approaches, including implicit and explicit solvent effects on crystal morphology and our modelling work done in this context is promising. I would like to thank the CEO of APC, Dr. Mark Barret for the opportunity to do this work, and my co-authors, Dr. Marko Ukrainczyk and Prof. Brian Glennon.