
A selection of case studies related to E-CAM’s pilot project activities focused on industrially oriented problems are published on this page. Some are short interviews with our postdoctoral researchers working on the related pilot projects. Others are success stories with clear analysis of the challenge, solution and benefit for the community. All material is available for download and is licensed under the Creative Commons Attribution 4.0 International License.
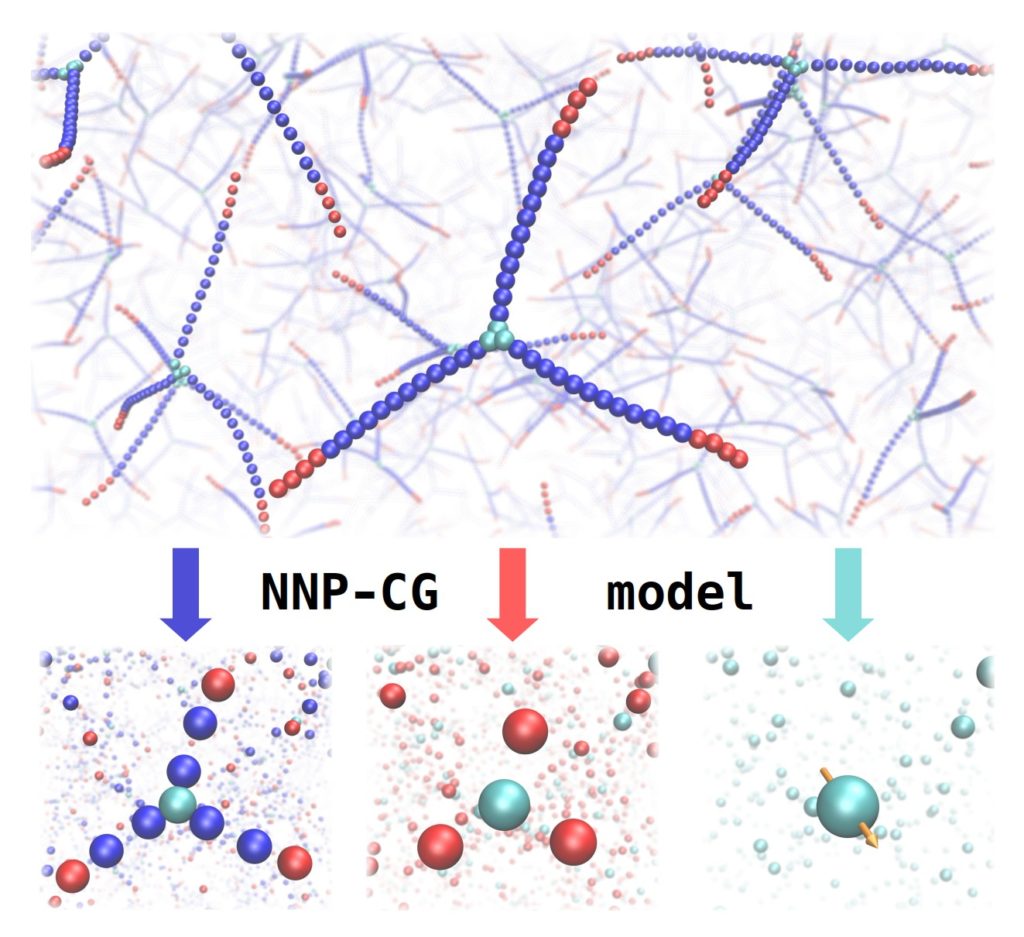
Implementation of High-Dimensional Neural Network Potentials
Dr. Andreas Singraber, VASP
[expand title=”Abstract”]In this conversation with Andreas Singraber, post-doc in E-CAM until last month, we will discover the ensemble of his work to expand the Neural Network Potential (NNP) Package n2p2 and to improve user accessibility to the code via the LAMMPS package. Andreas will talk about new tools that he developed during his E-CAM pilot project, that can provide valuable input for future developments of NNP based coarse-grained models. He will describe how E-CAM has impacted his career and led him to recently integrate a software company as a scientific software engineer.[/expand]

Calculations for Applications in Photovoltaic Devices
Dr. David Lopez, Universidad de Córdoba, Spain
[expand title=”Abstract”] The need to find easily renewable and environmentally friendly energy sources alternative to the traditional fossil fuels is nowadays a global quest. The solar energy is a promising candidate and organic solar cells (OSCs) have attracted attention. In this collaboration with Merck, E-CAM scientists have used electronic structure calculations to study how a key magnitude – the HOMO-LUMO band gap – changes with respect to the molecular disposition of the donor-acceptor molecule pair.[/expand]

[expand title=”Abstract”] In a recent paper [npj Computational Materials (2020) 6:66], researchers from the Centres of Excellence E-CAM and MaX[www.max-centre.eu], and the centre for Computational Design and Discovery of Novel Materials NCCR MARVEL[https://nccr-marvel.ch/], have proposed a new procedure for automatically generating Maximally-Localised Wannier functions (MLWFs) for high-throughput frameworks. The methodology and associated software can be used for hitherto difficult cases of entangled bands, and allows the electronic properties of a wide variety of materials to be obtained starting only from the specification of the initial crystal structure, including insulators, semiconductors and metals. Industrial applications that this work will facilitate include the development of novel superconductors, multiferroics, topological insulators, as well as more traditional electronic applications. [/expand]

The development of the GC-AdResS scheme
Dr. Christian Krekeler, Freie Universität Berlin, Germany
[expand title=”Abstract”] GC-AdResS is a technique that speeds up computations without loss of accuracy for key system properties by dividing the simulation box into two or more regions having different levels of resolution, for instance a high resolution region where the molecules of the system are treated at an atomistic level of detail, and other regions where molecules are treated at a coarse grained level, and transition regions where a weighted average of the two resolutions is used. The goal of the E-CAM GC-AdResS pilot project was to eliminate the need of a transition region so as to significantly improve performance, and to allow much greater flexibility. For example, the low resolution region can be a particle reservoir (ranging in detail from coarse grained to ideal gas particles) and a high resolution atomistic region with no transition region, as was needed hitherto. The only requirement is that the two regions can exchange particles, and that a corresponding “thermodynamic” force is computed self-consistently, which it turns out is very simple to implement.[/expand]

Mesoscale simulation of billion atom complex systems using thousands of GPGPUS’s
Dr. Jony Castagna, Science and Technology Facilities Council, UK

Mesoscale models for polarisable solvents: application to oil-water interfaces
Dr. Silvia Chiacchiera, Science and Technology Facilities Council, UK
[expand title=”Abstract”]
Water is a polar liquid and has a dielectric permittivity much higher than typical apolar liquids, such as light oils. This strong dielectric contrast at water-oil interfaces affects electrostatics and is important, for example, to include these effects to describe biomolecular processes and water-oil mixtures involving surfactants, as detergents. In this pilot project, developed in collaboration with Unilever and Manchester University, we have proposed and analysed a class of polarisable solvent models to be used in Dissipative Particle Dynamics (DPD), a coarse-grained particle-based simulation method commonly used in various industrial sectors. Related software modules for the DL_MESO package have also been developed.
[/expand]

Designing control pulses for superconducting qubit systems with local control theory
Dr. Momir Mališ, École Polytechnique Fédérale de Lausanne, Switzerland
[expand title=”Abstract”]
A quantum logic gate is one of the key components of the quantum computer, and designing an effective quantum universal gate is one of the major goals in the development of quantum computers. We have developed a software based on local control theory to design efficient state preparation control pulses for universal quantum gates which drive full population transfer between qubit states.
[/expand]

Dr. Hideki Kobayashi, Max Planck Institut für Polymerforschung, Germany
[expand title=”Abstract”]
The ability to accurately determine and predict properties of newly developed polymer materials is highly important to researchers and industry, but at the same time represents a significant theoretical and computational challenge. We have developed a novel multiscale simulation method based on the hierarchical equilibration strategy, which significantly decreases the equilibrium properties calculation time while satisfying the thermodynamic consistency. A number of E-CAM modules was developed and implemented in he ESPResSo++ software package.
[/expand]

The simulation of metal ions in protein-water systems using machine learning
Dr. Francesco Fracchia, Scuola Normale Superiore di Pisa, Italy
[expand title=”Abstract”]
One quarter to one third of all proteins require metals to function but the description of metal ions in standard force fields is still quite primitive. In this case study and interview an E-CAM project to develop a suitable parameterisation using machine learning is described. The training scheme combines classical simulation with electronic structure calculations to produce a force field comprising standard classical force fields with additional terms for the metal ion-water and metal ion-protein interactions. The approach allows simulations to run as fast as standard molecular dynamics codes, and is suitable for efficient massive parallelism scale-up.
[/expand]
To follow up with new case studies subscribe to our newsletter and keep an eye on our latest news on the website and on Twitter.
