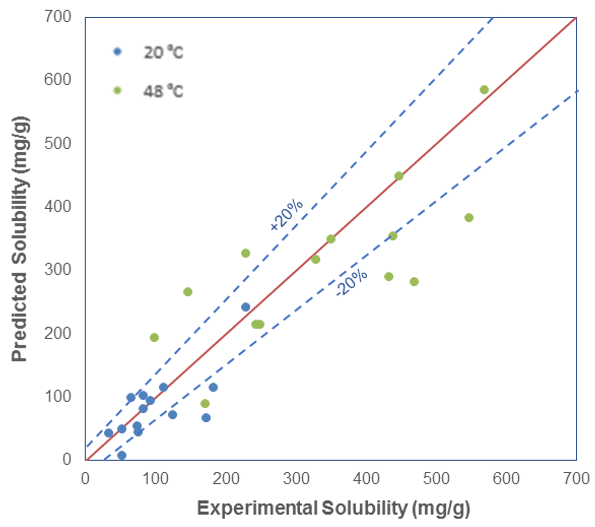
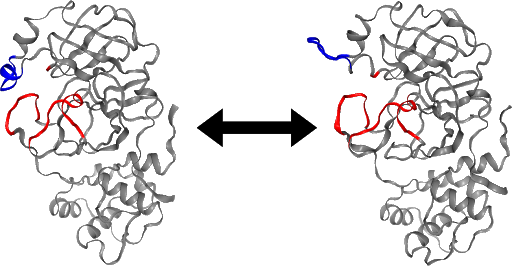
EKHAM the Comics What are “simulations” in advanced research? Is High Performance Computing the Holy Grail of scientific simulations? Let’s find out together through this unique Comic book Read more E-CAM A path to extreme-scale computing for industry and academia About us Keep up to date with the latest news from E-CAM Sign up to our quarterly newsletter December 2020 Newsletter E-CAM Survey of Application Software Fill in survey European Centre of Excellence Supporting HPC simulations in industry and academia through software development, training and discussion in simulation and modeling HIGHLIGHTS FROM E-CAM Quick Access Links E-CAM Issue of Comics&Science A Comic book about simulation, modelling and HPC. Case Studies Case studies and success stories related to pilot projects focused on industrially oriented problems Software Repository Access the software modules that have been documented by E-CAM Attend an event Our full calendar of events, that are part of the CECAM programme Online Training Portal Access the content captured at our ESDWs Scientific Publications Dissemination of E-CAM results in international peer-reviewed journals E-CAM's stories Interviews, opinion pieces, success stories, case studies. Modules of the month What is a module? Access our software repositories Comics & Science ? The E-CAM issue: an experiment in dissemination October 28, 2020 March 26, 2021 E-CAM Final Report June 10, 2021 May 31, 2022 Issue 16 – April 2021 April 13, 2021 April 13, 2021 Challenges to Industry of drug substance development March 30, 2021 March 31, 2021 Development of an HTC-based, scalable committor analysis tool in OpenPathSampling opens avenues to investigate enzymatic mechanisms linked to Covid-19 March 29, 2021 April 12, 2021 Proof of concept : recognition as a disruptive technology March 29, 2021 March 29, 2021 Upcoming E-CAM events Recent Posts Comics & Science ? The E-CAM issue: an experiment in dissemination October 28, 2020 March 26, 2021 E-CAM Final Report June 10, 2021 May 31, 2022 Issue 16 – April 2021 April 13, 2021 April 13, 2021 Challenges to Industry of drug substance development March 30, 2021 March 31, 2021 Development of an HTC-based, scalable committor analysis tool in OpenPathSampling opens avenues to investigate enzymatic mechanisms linked to Covid-19 March 29, 2021 April 12, 2021 Proof of concept : recognition as a disruptive technology March 29, 2021 March 29, 2021