Development of an HTC-based, scalable committor analysis tool in OpenPathSampling opens avenues to investigate enzymatic mechanisms linked to Covid-19
The E-CAM HPC Centre of Excellence and a PRACE team in Wroclaw have teamed up to develop High Throughput Computing (HTC) based tools to enable the computational investigation of reaction mechanisms in complex systems. These tools might help us gain understanding of enzymatic mechanisms used by the SARS-CoV-2 main protease [1]
Studying reaction mechanisms in complex systems requires powerful simulations. Committor analysis is a powerful, but computationally expensive tool, developed for this purpose. An alternative, less expensive option, consists in using the committor to generate initial trajectories for transition path sampling. In this project, the main goal was to integrate the committor analysis methodology with an existing software application, OpenPathSampling [2,3] (OPS), that performs many variants of transition path sampling (TPS) and transition interface sampling (TIS), as well as other useful calculations for rare events. OPS is performance portable across a range of HPC hardware and hosting sites..
The Committor analysis is essentially an ensemble calculation that maps straightforwardly to an HTC workflow, where typical individual tasks have moderate scalability and indefinite duration. Since this workflow requires dynamic and resilient scalability within the HTC framework, OPS was coupled to E-CAM’s HTC library jobqueue_features[4] that leverages the Dask [5, 6] data analytics framework and implements support for the management of MPI-aware tasks.
The HTC library jobqueue_features proved to be resilient and to scale extremely well, meaning it can handle a very high number of simultaneous tasks: a stress test showed it can scale out to 1M tasks on all available architectures. OPS was expanded and its integration with the jobqueue features library was made trivial. In its current state, OPS can now almost seamlessly transition from use on a personal laptop to some of the largest HPC sites in Europe.
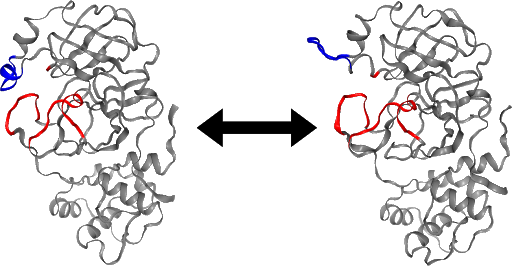
Integrating OPS and the HTC library resulted in an unprecedented parallelised committor simulation capability. These tools are currently being implemented for a committor simulation of the SARS-CoV-2 main protease. An initial analysis of the stable states, based on a long trajectory provided by D.E. Shaw Research [7] suggests that a loop region of the protein may act as a gate to the active site (Figure). This conformational change may regulate the accessibility of the active site of the main protease, and a better understanding of its mechanism could aid drug design.
The committor simulation can be used to explore the configuration space (taking more initial configurations), or to improve the accuracy of the calculated committor value (running more trajectories per configuration). Altogether, such data will provide insight into the dynamics of the protease loop region and the mechanism of its gate-like activity. In addition, the trajectories generated by the committor simulation can also be used as initial conditions for further studies using the transition path sampling approach.
References
[1] Milosz Bialczak, Alan O’Cais, Mariusz Uchronski, & Adam Wlodarczyk. (2020). Intelligent HTC for Committor Analysis. http://doi.org/10.5281/zenodo.4382017
[2] David W.H. Swenson, Jan-Hendrik Prinz, Frank Noé, John D. Chodera, and Peter G. Bolhuis. “OpenPathSampling: A flexible, open framework for path sampling simulations. 1. Basics.” J. Chem. Theory Comput. 15, 813 (2019). https://doi.org/10.1021/acs.jctc.8b00626
[3] David W.H. Swenson, Jan-Hendrik Prinz, Frank Noé, John D. Chodera, and Peter G. Bolhuis. “OpenPathSampling: A flexible, open framework for path sampling simulations. 2. Building and Customizing Path Ensembles and Sample Schemes.” J. Chem. Theory Comput. 15, 837 (2019). https://doi.org/10.1021/acs.jctc.8b00627
[4] Alan O’Cais, David Swenson, Mariusz Uchronski, and Adam Wlodarczyk. Task Scheduling Library for Optimising Time-Scale Molecular Dynamics Simulations, August 2019.
[5] Dask Development Team. Dask: Library for dynamic task scheduling, 2016.
[6] Matthew Rocklin. Dask: Parallel computation with blocked algorithms and task scheduling. In Kathryn Hu and James Bergstra, editors, Proceedings of the 14th Python in Science Conference, pages 130 – 136, 2015.[7] No specific author. Long trajectory provided by D.E. Shaw Research. https://www.deshawresearch.com/downloads/download_trajectory_sarscov2.cgi/, 2020. [Online; accessed 22-Oct-2020].