To study the properties of polymer melts by numerical simulations, equilibrated configurations must be prepared. However, the relaxation time for high molecular weight polymer melts is huge and increases, according to reptation theory, with the third power of the molecular weight. Hence, an effective method for decreasing the equilibration time is required. The hierarchical strategy pioneered in Ref. [1] is a particularly suitable way to do this. The present module provides a part of that method.
To decrease the relaxation time, microscopic monomers are coarse-grained (CG) by mapping each subchain with 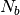 monomers onto a soft blob. The CG system is then characterized by a much lower molecular weight and thus is equilibrated quickly. The present module provides a python script which performs this coarse-graining procedure. The implementation details can be seen in the module’s documentation on our software Library here. This module is part of a set of codes that together implement the Hierarchical Equilibration strategy of Ref. [1], in the ESPResSO++ [2] (for the complete list of modules, see here under ESPResSO++).
monomers onto a soft blob. The CG system is then characterized by a much lower molecular weight and thus is equilibrated quickly. The present module provides a python script which performs this coarse-graining procedure. The implementation details can be seen in the module’s documentation on our software Library here. This module is part of a set of codes that together implement the Hierarchical Equilibration strategy of Ref. [1], in the ESPResSO++ [2] (for the complete list of modules, see here under ESPResSO++).
Practical application and exploitation of the code
The development of a multiscale method for polymer blends and block copolymers is fundamentally new and needs to be based on first-principles theory. This is therefore an intellectual challenge in its own right. Furthermore, this paves the way to analyze the physical properties of novel composite materials that attract the attention of industrial companies. Such materials may be promising ingredients of new products like e.g. efficient and environment-friendly car tires. The implementation of the Hierarchical Equilibration strategy in the ESPResSO++ package is a step towards achieving this goal. In particular, the practical application of this strategy is the E-CAM pilot project in collaboration with Michelin aimed at studying the Rheological Properties of New Composite Materials.
E-CAM deliverables D4.2 and D4.3 contain more information on the suite of programs developed under this pilot project.
[1] Zhang, G., Moreira, L. A., Stuehn, T., Daoulas, K. C., and Kremer, K., Equilibration of High Molecular Weight Polymer Melts: A Hierarchical Strategy, ACS Macro Lett., 3, 198-203 (2014)
[2] ESPResSo++ is the “Extensible Software Package for Research in Soft Matter based upon C++”, a general-purpose simulation package for soft-matter research, mainly developed at the Max Planck Institute for Polymer Research Mainz. It is freely available under the GNU Public License. http://www.espresso-pp.de/

