PIM_wd: Module for sampling of the quantum Wigner distribution
The PIM_wd module implements the exact quantum Wigner probability distribution function sampling algorithm of the Phase Integration Method [1], and is the main subroutine for the quantum correlation function calculations in the PaPIM code. The module samples the thermal Wigner density using a generalised Monte Carlo scheme for sampling phase space points. The scheme combines the Penalty [2] and Kennedy [3] algorithms to sample noisy probability densities. This is necessary because the estimator of the quantum thermal density is not known analytically but must be computed via a statistical average affected by uncertainty. The sampled points are the basis for the calculation of time-independent and time-dependent system observables.
The module was developed as the main component of the PaPIM code, but also as a standalone subroutine that can be easily implemented in other methods (e.g. the whole family of so-called linearised approximations of quantum dynamics) for which phase space sampling of the exact quantum Wigner probability distribution is required. Because the Phase Integration Method samples a set of independent phase space points, independent instances of the PIM_wd module can be run in parallel in order to parallelise the phase space sampling. In the PaPIM package, the parallelisation is accomplished using MPI, which has proved to provide good scalability of the PaPIM code. The module will also be adapted for HTC capabilities.
Practical application and exploitation of the code
The code has been extensively used for the calculation of the infrared absorption spectrum of CH5+ in the gas phase. [4] This highly flexible molecule is considered a standard benchmark of approximate quantum methods, and has experimental interest, for example, in the context of green chemistry.
This module is part of the modules in deliverable D3.3 which were developed during the E-CAM ESDW7.
[1] M. Monteferrante, S. Bonella, G. Ciccotti Quantum dynamical structure factor of liquid neon via a quasiclassical symmetrized method J. Chem. Phys. 138 (2013) 054118
[2] D. M. Ceperley, M. Dewing The penalty method for random walks with uncertain energies J. Chem. Phys. 110 (1999) 9812
[3] A. D. Kennedy, J. Kuti Noise without Noise: A New Monte Carlo Method Phys. Rev. Lett. 54 (1985) 2473
[4] O. Asvany, P. K. P, B. Redlich, I. Hegemann, S. Schlemmer, D. Marx Understanding the infrared spectrum of bare CH5+ Science 309 (2005) 1219

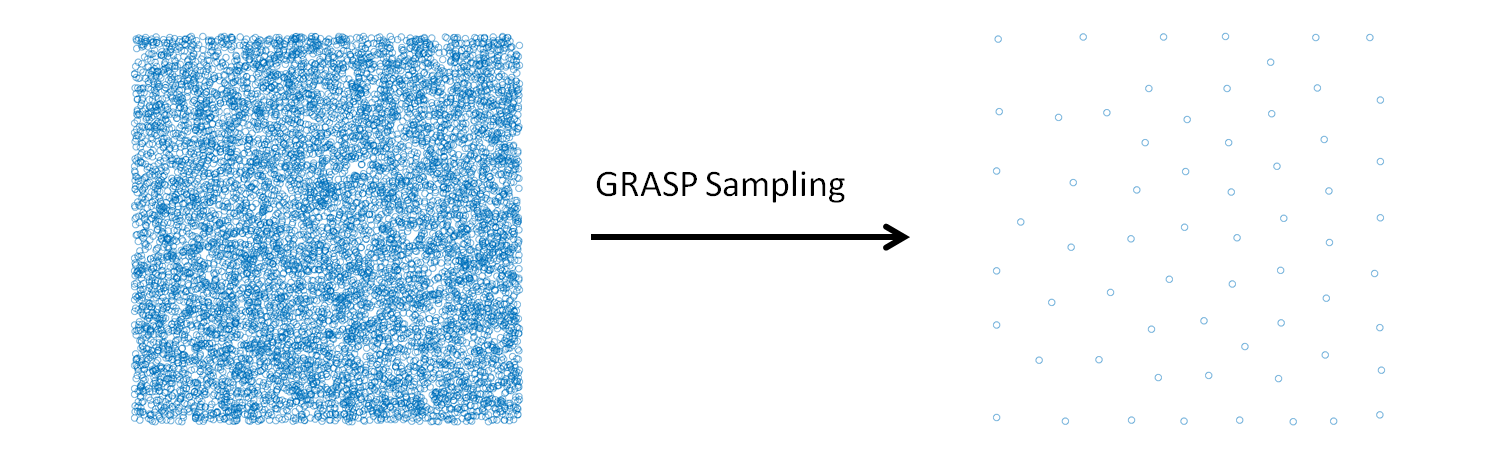
 monomers onto a soft blob. The CG system is then characterized by a much lower molecular weight and thus is equilibrated quickly. The present module provides a python script which performs this coarse-graining procedure. The implementation details can be seen in the module’s documentation on our software Library
monomers onto a soft blob. The CG system is then characterized by a much lower molecular weight and thus is equilibrated quickly. The present module provides a python script which performs this coarse-graining procedure. The implementation details can be seen in the module’s documentation on our software Library