

Issue 7 – January 2018
E-CAM Newsletter of January 2018
Get the latest news from E-CAM, sign up for our quarterly newsletter.
The simulation of metal ions in protein-water systems using machine learning: An E-CAM case study and conversation
With Dr. Francesco Fracchia, Scuola Normale Superiore di Pisa
Interviewer: Dr. Donal Mackernan, University College Dublin
Abstract
One quarter to one third of all proteins require metals to function but the description of metal ions in standard force fields is still quite primitive. In this case study and interview an E-CAM project to develop a suitable parameterisation using machine learning is described. The training scheme combines classical simulation with electronic structure calculations to produce a force field comprising standard classical force fields with additional terms for the metal ion-water and metal ion-protein interactions. The approach allows simulations to run as fast as standard molecular dynamics codes, and is suitable for efficient massive parallelism scale-up.
GRASP Sampling – a module to build a representative data set for a fitting procedure
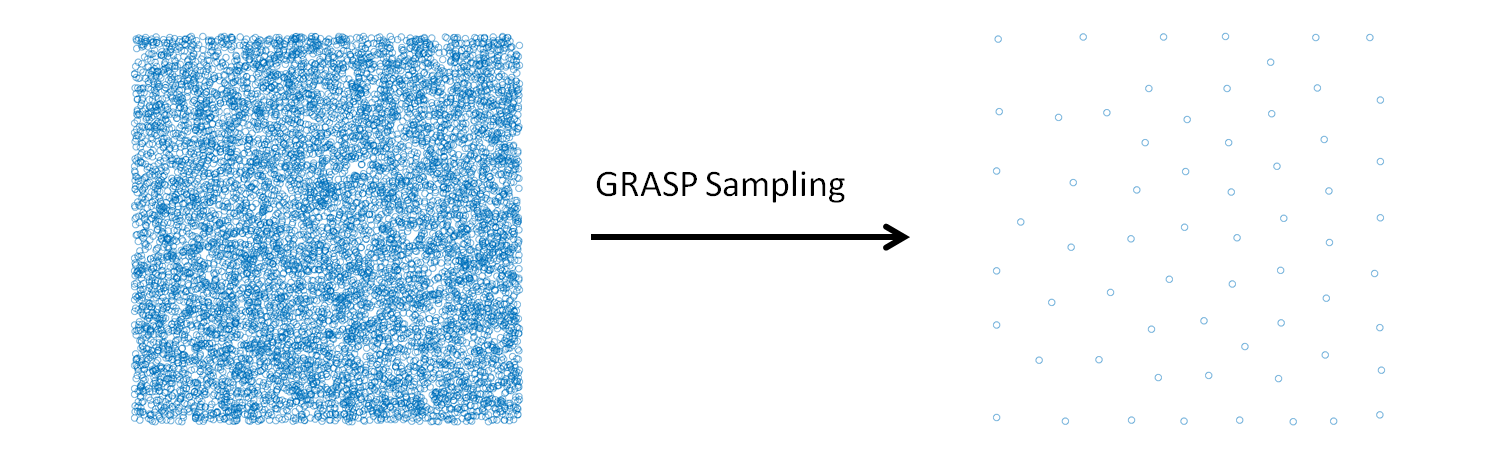
GRASP_sampling performs a stratified sampling of the configurations, described by vectors, of a system to build a representative training set in a fitting procedure. Given a list of candidate configurations, and selected the size (N) of the training set required, the module executes the combinatorial optimization that maximizes the following dissimilarity score (DS) among the elements of the training set:
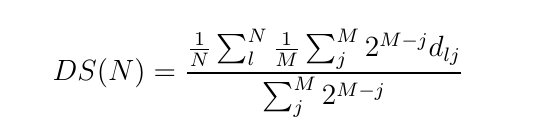
In this formula, the j-th configuration in the sum is the j-th nearest one to the l-th configuration and dij is the Euclidean distance between the l-th and j-th configurations. M is the number of the nearest configurations considered in the score. The exponential weight makes the score near independent from the particular value of M, if it is larger than 4-6.
The combinatorial optimization that maximizes the dissimilarity score is performed using the greedy randomized adaptive search procedure[1] (GRASP) algorithm. A stratified sampling can be performed without a combinatorial optimization using classical statistical techniques (for example Latin hypercube sampling), the GRASP sampling becomes useful when the selection is restricted to a predeterminated set of configurations, generated or sampled with specific internal constrains. This is the case of the molecular configurations generated in a molecular dynamics simulation.
The complete module documentation, including a link to the source code, can be found in our repository here.
Motivation and exploitation
The application of the GRASP algorithm to perform a stratified sampling is described in a recent publication [2] by the E-CAM partners at Scuola Normale Superiore (SNS), that we previously reported here.
The motivation behind this software module is the pilot project with industry “Quantum Mechanical Parameterisation of Metal Ions in Proteins” sustained by an E-CAM postdoctoral researcher from SNS.
[1] Feo, T. A.; Resende, M. G. Greedy randomized adaptive search procedures. J. Glob. Optim. 1995, 6, 109−133
[2] Francesco Fracchia, Gianluca Del Frate, Giordano Mancini, Walter Rocchia, and Vincenzo Barone, Force Field Parametrization of Metal Ions from Statistical Learning Techniques, J. Chem. Theory Comput. 2018, 14, 255−273
From Rational Design of Molecular Biosensors to Patent and potential Start-up
Dr. Donal Mackernan, University College Dublin
Abstract
The power of advanced simulation combined with statistical theory , experimental know-how and high performance computing is used to design a protein based molecular switch sensor with remarkable sensitivity and significant industry potential. The sensor technology has applications across commercial markets including diagnostics, immuno-chemistry, and therapeutics.
New publication using the GC-AdResS molecular dynamics technique
The publication “Probing spatial locality in ionic liquids with the grand canonical adaptive resolution molecular dynamics technique (GC-AdResS)“ by the Theoretical and Mathematical Physics in Molecular Simulation group of the Freie Universität Berlin, lead by Prof.Luigi Delle Site, E-CAM partner, describes the use of the GC-AdResS molecular dynamics technique to test the spatial locality of the ionic liquid 1-ethyl 3-methyl imidazolium chloride liquid. The main aspect of GC-AdResS is the possibility to couple two simulation boxes together and combine the advantages of classical atomistic simulations with those from coarse gained simulations.
The publication post-print version is open access and can be downloaded directly from the Zenodo repository here. The publisher AIP version can be found at http://aip.scitation.org/doi/10.1063/1.5009066.
E-CAM currently runs a pilot project on the development of the GC-AdResS scheme and one of its goals is to develop a library or recipe with which GC-AdResS can be implemented in any MD Code. The current focus is to adjust the implemented version of GC-AdResS in GROMACS. The long-term goal of this project is to promote and stimulate the community to use it as a tool for multiscale simulations and analysis. More information about this pilot project can be found here.
Article
Title: Probing spatial locality in ionic liquids with the grand canonical adaptive resolution molecular dynamics technique
Authors: B. Shadrack Jabes, C. Krekeler, R. Klein and L. Delle Site
Abstract: We employ the Grand Canonical Adaptive Resolution Simulation (GC-AdResS) molecular dynamics technique to test the spatial locality of the 1-ethyl 3-methyl imidazolium chloride liquid. In GC-AdResS, atomistic details are kept only in an open sub-region of the system while the environment is treated at coarse-grained level; thus, if spatial quantities calculated in such a sub-region agree with the equivalent quantities calculated in a full atomistic simulation, then the atomistic degrees of freedom outside the sub-region play a negligible role. The size of the sub-region fixes the degree of spatial locality of a certain quantity. We show that even for sub-regions whose radius corresponds to the size of a few molecules, spatial properties are reasonably reproduced thus suggesting a higher degree of spatial locality, a hypothesis put forward also by other researchers and that seems to play an important role for the characterization of fundamental properties of a large class of ionic liquids.
The Journal of Chemical Physics 148, 193804 (2018)
E-CAM program of events 2018 is out

Check out our program of events for this year, running from April 2018 to February 2019:
See the workshop details to learn how to apply. E-CAM events are part of the annual CECAM flagship program, and are hosted at the different CECAM Nodes locations.
E-CAM runs three types of events every year:
- Scoping workshops (SCOWs)
- State-of-the-art workshop (SAWs)
- Extended Software Development Workshops (ESDWs)
For their definition see here. If you require any further information contact us at info@e-cam2020.eu
New article is out: “Force Field Parametrization of Metal Ions from Statistical Learning Techniques”
This paper from E-CAM partners working in Scuola Normale Superiore (Pisa, Italy) describes a novel statistical procedure, developed to optimize the parameters of non-bonded force fields of metal ions in soft matter. The paper is open access and can be downloaded directly from ACS’s page at http://pubs.acs.org/doi/10.1021/acs.jctc.7b00779.
This work was performed in the context of the E-CAM pilot project on Quantum Mechanical Parameterisation of Metal Ions in Proteins, which is a collaboration with BiKi Technologies. The list of software modules associated to the pilot project (and this publication) can be found here.
Article
Title: Force Field Parametrization of Metal Ions from Statistical Learning Techniques
Authors: Francesco Fracchia, Gianluca Del Frate, Giordano Mancini, Walter Rocchia, and Vincenzo Barone
Abstract: A novel statistical procedure has been developed to optimize the parameters of non-bonded force fields of metal ions in soft matter. The criterion for the optimization is the minimization of the deviations from ab initio forces and energies calculated for model systems. The method exploits the combination of the linear ridge regression and the cross-validation techniques with the di˙erential evolution algorithm. Wide freedom in the choice of the functional form of the force fields is allowed since both linear and non-linear parameters can be optimized. In order to maximize the information content of the data employed in the fitting procedure, the composition of the training set is entrusted to a combinatorial optimization algorithm which maximizes the dissimilarity of the included instances. The methodology has been validated using the force field parametrization of five metal ions (Zn2+, Ni2+, Mg2+, Ca2+, and Na+) in water as test cases.
Geomoltools: A set of software modules to easily manipulate molecular geometries
Geomoltools is a set of eight pre- and post-treatment Fortran codes that can be used to easily manipulate molecular geometries, allowing to minimize the average energy obtained for a range of internuclear distances for the dimers of each element, and decrease the computational cost of a DFT calculation.
The set of codes are:
- mol2xyz: converts a .mol file into an ordered .xyz file
- pastemol: joins two .xyz files
- movemol: translates and aligns the molecule with some predefined axes
- stackmol: generates (manually or randomly) different stacking arrangements between two molecules
- geodiff: compares the internal coordinates of two molecules
- xyz2zmt_s: converts the cartesian coordinates contained in a .xyz file into Z-matrix (2 possible formats)
- zmt2xyz_s: converts a Z-matrix (from 2 possible formats) into cartesian coordinates
- ucubcellgen: calculates the vectors of a unit cell given some atomic coordinates.
Modules source codes can be found here. For a detailed explanation of the main programs, please have a look to this file. A complete tutorial on how to use the different codes from the package Geomoltools in order to manipulate (rotate, translate, join, pack, convert, etc.) molecular geometries, can be found at this address.
Motivation and exploitation
These modules have been used to study the stacking arrangements of acceptor:donor molecules for organic photovolatics polymers by high-throughput computation with the SIESTA code. This set of codes are available under the GNU General Public License (GPL) version 2.