
GRASP Sampling – a module to build a representative data set for a fitting procedure
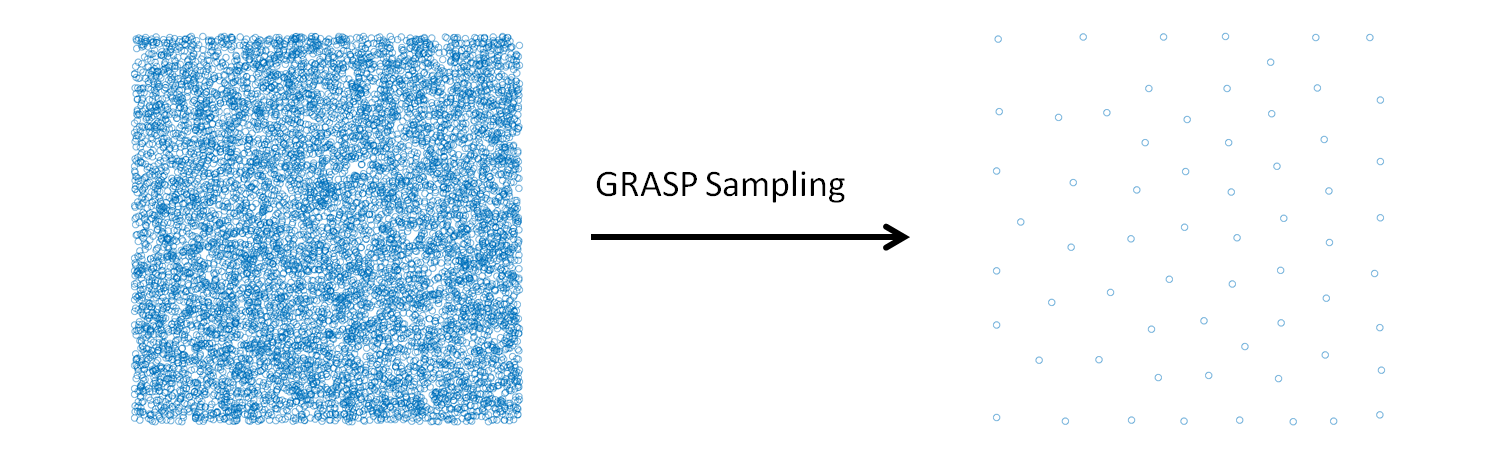
GRASP_sampling performs a stratified sampling of the configurations, described by vectors, of a system to build a representative training set in a fitting procedure. Given a list of candidate configurations, and selected the size (N) of the training set required, the module executes the combinatorial optimization that maximizes the following dissimilarity score (DS) among the elements of the training set:
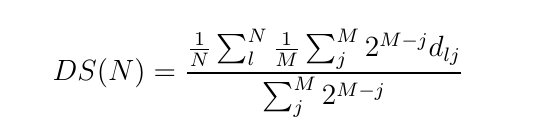
In this formula, the j-th configuration in the sum is the j-th nearest one to the l-th configuration and dij is the Euclidean distance between the l-th and j-th configurations. M is the number of the nearest configurations considered in the score. The exponential weight makes the score near independent from the particular value of M, if it is larger than 4-6.
The combinatorial optimization that maximizes the dissimilarity score is performed using the greedy randomized adaptive search procedure[1] (GRASP) algorithm. A stratified sampling can be performed without a combinatorial optimization using classical statistical techniques (for example Latin hypercube sampling), the GRASP sampling becomes useful when the selection is restricted to a predeterminated set of configurations, generated or sampled with specific internal constrains. This is the case of the molecular configurations generated in a molecular dynamics simulation.
The complete module documentation, including a link to the source code, can be found in our repository here.
Motivation and exploitation
The application of the GRASP algorithm to perform a stratified sampling is described in a recent publication [2] by the E-CAM partners at Scuola Normale Superiore (SNS), that we previously reported here.
The motivation behind this software module is the pilot project with industry “Quantum Mechanical Parameterisation of Metal Ions in Proteins” sustained by an E-CAM postdoctoral researcher from SNS.
[1] Feo, T. A.; Resende, M. G. Greedy randomized adaptive search procedures. J. Glob. Optim. 1995, 6, 109−133
[2] Francesco Fracchia, Gianluca Del Frate, Giordano Mancini, Walter Rocchia, and Vincenzo Barone, Force Field Parametrization of Metal Ions from Statistical Learning Techniques, J. Chem. Theory Comput. 2018, 14, 255−273



