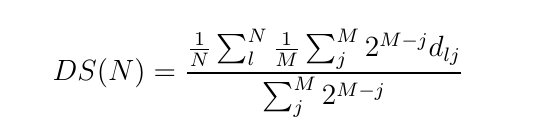Improving I/O of DL_MESO_DPD files using SIONlib
This module implements the SIONlib library to optimize the I/O (writing/reading) of the trajectory files generated by DL_MESO_DPD, the Dissipative Particle Dynamics (DPD) code from the DL_MESO package. SIONlib is a library for writing and reading binary data to/from several thousands of processors into one or a small number of physical files. For parallel access to files, only the open and close functions are collective, while the writing and reading of files can be done asynchronously. [1] In DL_MESO_DPD’s last release (version 2.6), the MPI version of DL_MESO_DPD generates multiple trajectory files, one for each MPI task. The interface with SIONlib optimizes the data writing so that just one physical file is produced from several MPI tasks. This drastic reduction in the number of output files is a benefit for the I/O of the code, and simplifies the maintenance of the output, especially for a large number of MPI tasks.
This module is part of the newly developed utilities for the DL_MESO_DPD code within the pilot project on Polarizable Mesoscale Models.
Practical application and exploitation of the code
The implementation of this module generates a single trajectory file (history.sion) in a parallel run of DL_MESO_DPD, instead of multiple (HISTORY) ones. Accordingly, analogous modifications have to be implemented in the post-processing utilities that read the HISTORY files. As an example, the changes were implemented in a formatting utility. Besides showing how to adapt the reading, this allows a robust check of the implementation, since the output is human readable, contains the full trajectories, and can be readily compared with outputs obtained using the standard version of DL_MESO_DPD.
The next released version of DL_MESO_DPD (in development) will tackle the writing of files differently, producing a single trajectory file from the start. However, the interface proposed here provides this feature to the users of version 2.6, and represents an alternative solution for the handling of the trajectories.
It should be noted that this implementation is meant to show the feasibility of the interfacing, not to deal with all the possible cases. Thus, the module’s functionality is restricted to the relevant case in which: i) the simulation is run in parallel using MPI, ii) a single SIONlib-type physical file is produced, and iii) the post-processing is done by a single process.
While SIONlib is optimized for a large number of MPI tasks, even the reduction from several output files to just one represents a benefit, for example when it comes to the maintenance of the simulation output.
[1] http://www.fz-juelich.de/ias/jsc/EN/Expertise/Support/Software/SIONlib/_node.html

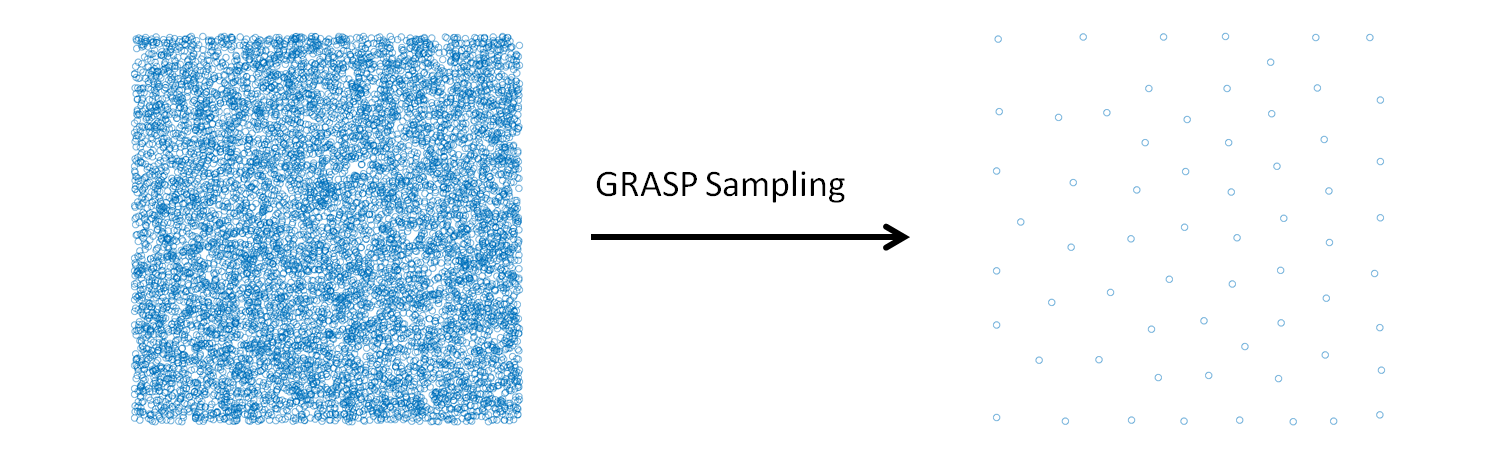
 monomers onto a soft blob. The CG system is then characterized by a much lower molecular weight and thus is equilibrated quickly. The present module provides a python script which performs this coarse-graining procedure. The implementation details can be seen in the module’s documentation on our software Library
monomers onto a soft blob. The CG system is then characterized by a much lower molecular weight and thus is equilibrated quickly. The present module provides a python script which performs this coarse-graining procedure. The implementation details can be seen in the module’s documentation on our software Library